US Pharm. 2014;39(8):34-38.
ABSTRACT: Opioid analgesics are considered the mainstay of pain management. The primary goal of analgesia is optimizing the patient’s comfort. However, dosing these agents may be complicated in patients with renal and/or hepatic impairment. Most recommendations are based on case reports; therefore, appropriate analgesic agent selection requires a thorough understanding of the drugs’ pharmacokinetics and side-effect profiles. This article will review opioid analgesics and their vital clinical pharmacokinetic considerations when treating patients with hepatic and/or renal insufficiency.
Pain is a condition affecting more Americans than diabetes, heart disease, and cancer combined, with an estimated incidence of 100 million people.1 Comorbid conditions such as hepatic and renal disease may complicate pain treatment with appropriate opioid analgesics.
EPIDEMIOLOGY AND MECHANISM OF ACTION
The majority of opioids are metabolized through phase I biotransformation via CYP450 enzymes 2D6 and 3A4.2 Hepatic impairment is a condition that may lead to increased opioid toxicity. This occurrence is secondary to the parent drug being inadequately converted to the metabolite for elimination. Consideration of both the parent compound and metabolite accumulation should also be accounted for in patients with renal insufficiency.
According to Davidson and Jhangri’s study involving 205 patients on hemodialysis (HD), up to one-half of patients receiving HD experienced chronic pain.3 Eighty-two percent of this patient population experienced moderate-to-severe pain, thereby justifying the need for stronger opioids.3 A total of 103 patients were experiencing chronic pain; 26.2% of these chronic pain patients were prescribed weak opioids, while only 10% were given strong opioids.4 Hence, understanding opioid pharmacokinetics is imperative to adequately control pain and minimize toxicity and adverse effects.
PHARMACOKINETICS IN ORGAN IMPAIRMENT
The pharmacokinetics of a drug depends on patient-related factors as well as on the drug’s chemical properties. Some patient-related factors (e.g., renal function, genetic makeup, sex, age) can be used to predict the pharmacokinetic parameters in a given population. Variability in these parameters—absorption, distribution, metabolism, and excretion—may occur in patients with hepatic and renal impairment. Vital aspects of opioid pharmacokinetics will be discussed in the subsequent sections.
Hepatic Impairment
Absorption: Patients with liver cirrhosis often develop gastritis, portal hypertensive gastropathy, or ulcers of the gastrointestinal (GI) tract.5 These conditions, coupled with delayed gastric emptying in cirrhotic patients, may lead to delayed drug absorption.5,6 As a result, it is suggested that patients with cirrhosis be prescribed immediate-release opioids rather than extended, sustained-release, or delayed-release formulations.
Distribution: Patients experiencing cirrhotic liver with ascites have an increased volume of distribution secondary to third spacing. Therefore, loading doses of many hydrophilic agents may warrant an increase (e.g., beta-lactams and digoxin).5 Due to other pharmacokinetic parameter considerations and the increased risk of adverse effects of hydrophilic opioids (e.g., morphine, oxycodone, hydromorphone), it is prudent to start with lower initial doses and titrate slowly to effect.7
Protein binding of opioids is another concept pertaining to distribution that warrants careful consideration. Opioids possessing a high protein-binding profile may have an increased free-drug level in patients with liver insufficiency because of their decreased production of alpha-1-acid glycoprotein and albumin. Hence, high protein-binding properties may lead to toxicity.8 Examples of highly protein-bound opioids are methadone (80%-90%) and buprenorphine (96%).7,9
Metabolism: The liver is the major site of biotransformation from parent opioid compounds to active or inactive metabolites. Tools, such as the Child-Pugh classification or the Model for End-Stage Liver Disease (MELD) score, are used for prognosis in patients with cirrhosis. Unfortunately, neither of these tools nor other endogenous markers are able to provide any assessment of hepatic clearance; therefore, an alternative approach in determining drug dosing is needed in the hepatic impairment patient population. Nonetheless, one possible way utilized in dosage guidance is classifying drugs by the extent to which the liver metabolizes them, a process known as the hepatic extraction ratio. This ratio ranges from 0 to 1, with 0 reflecting the inability of the liver to metabolize the drug and a ratio of 1 reflecting the ability to metabolize the entire drug via first pass. Medications with a high extraction ratio are identified as having an extraction ratio of >0.7, while an intermediate is between 0.3 and 0.7 and a low is <0.3.8 Morphine and fentanyl are categorized as high extraction ratio agents, while methadone is of a low extraction ratio (TABLE 1).
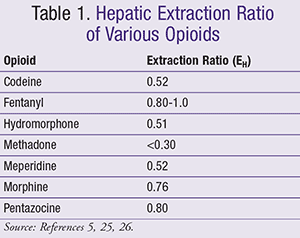
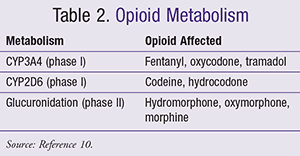
Development of portal-systemic shunts occurs in patients with cirrhosis, leading to decreased blood flow and subsequently halting drug metabolism by the liver.8 A conservative approach must be considered, assuming high bioavailability, when administering oral opioids with a high extraction ratio. These highly hepatically metabolized agents are bypassing first-pass metabolism as a result of the shunts.8 In addition, determining the primary pathway for metabolism, whether phase I reactions (CYP450) or phase II (i.e., glucuronidation), is vital when treating this patient population with opioids. CYPP450-mediated reactions are altered more by hepatic impairment, whereas phase II reactions are less affected (TABLE 2).10 Moreover, although certain opioids, such as morphine, primarily go through a phase II reaction, it is vital to consider metabolites leading to toxicity, in addition to the high extraction ratio profile.
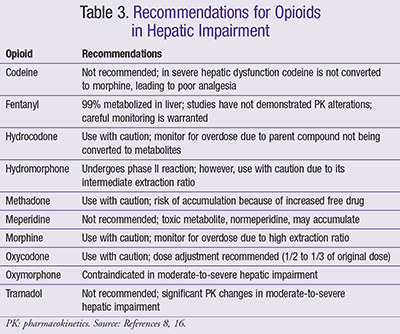
Excretion: Mild-to-moderate liver disease may lead to renal impairment.7 Specific recommendations for patients with hepatic insufficiency are summarized in TABLE 3.
Renal Impairment
Three mechanisms influencing renal excretion of opioids exist: glomerular filtration, tubular secretion, and tubular reabsorption. Estimations of the glomerular filtration rate (GFR) are used to predict renal excretion of medications, due to the lack of feasibility in estimating tubular secretion and reabsorption. Renal adjustment for medications may also be based on creatinine clearance (CrCl).
Formulas, such as the Cockcroft-Gault equation, that aid in predicting renal clearance, especially in the hepatic insufficiency population, may yield poor estimation of an agent’s renal clearance. This is due to the variability in muscle mass and the decreased conversion of creatine to creatinine.7 Nevertheless, patients with renal insufficiency are commonly treated with opioids. Approximately one-third of patients experiencing CrCl of <50 mL/min are administered an opioid for pain.4
The following section will review clinical considerations when selecting an opioid in the renal impairment and dialysis patient population.
Pharmacologic Approach in Renal Impairment and Dialysis
Morphine: Morphine, which was invented in 1804, is among the oldest and most studied drugs compared to other opioid analgesics.11 Greater than 50% of the parent compound is metabolized to morphine-3-glucuronide (M3G), while 10% is converted to morphine-6-glucuronide (M6G).9 In rodent studies, M3G has been proven to cause neuronal excitation despite the absence of analgesic effects.9
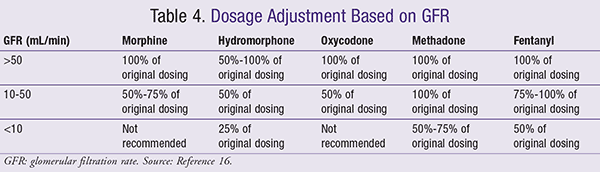
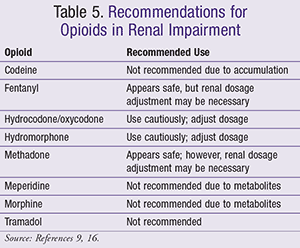
Conversely, M6G, a mu receptor agonist, possesses analgesic properties.2 In renal insufficiency, the clearance of the parent compound (morphine) is not significantly affected; however, M3G and M6G accumulation does occur.9 Therefore, it is recommended that morphine be used cautiously in patients with severe renal insufficiency (TABLES 4 and 5).9
Drugs with a low volume of distribution (Vd) and low protein binding are hydrophilic, and possess low-molecular-weight properties that are recognized as dialyzable agents. Morphine is known to have a low Vd and is water-soluble; however, a form of its M6G metabolite is lipophilic.9 It was hypothesized that M6G did not undergo complete dialysis removal, thus reequilibrating back into the central nervous system (CNS), leading to excessive sedation; this process is described in a case report by Angst et al as the rebound phenomenon post dialysis.12 As a result, it is recommended to avoid use of morphine in dialysis patients.
Hydromorphone: Approximately 37% of hydromorphone undergoes glucuronidation to produce hydromorphone-3-glucuronide (H3G).9,11 Similar to M3G, H3G has the potential to cause neuronal excitation.2 According to a study conducted by Lee et al, 29 patients with elevated urea and creatinine were compared to 26 patients with normal renal function who were switched to hydromorphone, mainly because of cognitive effects from a previous opioid.13 Eighty percent of patients had improvement in cognitive effects, and 55% reported improvement in pain.13 The authors concluded that hydromorphone was a safe and effective agent in patients with end-stage renal failure. Practitioners should still exercise caution when prescribing hydromorphone due to its risk of accumulation.
In an open, parallel-group, single-dose study involving 23 patients, 4-mg of immediate-release hydromorphone (Dilaudid IR) was administered. The results of the study evidenced an increase in the AUC corresponding with worsening of renal function.14 Two of the 23 trial patients underwent a 4-hour hemodialysis session and were noted to have experienced a 40% reduction in the parent compound.14
More data are needed in the setting of dialysis with hydromorphone. Hydromorphone is water-soluble and has low-molecular-weight properties.9, 11 Overall, it is recommended to closely monitor patients with moderate renal insufficiency during dose titration, while increasing the dosing interval in severe renal insufficiency patients.
Oxycodone: Oxycodone is metabolized to noroxycodone via CYP3A4 and oxymorphone via CYP2D6.15 Oxymorphone is an active metabolite with negligible plasma levels, while noroxycodone is a weaker opioid than the parent compound.2,16 Uremic patients were found to experience an increased half-life of oxycodone despite its 8% to 14% elimination rate as the parent compound.9,17 Caution should be exercised when dosing oxycodone in patients with renal insufficiency, as dose adjustments may be warranted (TABLE 4).16
When compared with hydromorphone, oxycodone was found to have a higher volume of distribution and be 50% protein-bound.9 According to Foral et al’s case report, a 41-year-old hemodialysis patient who received several doses of oxycodone/acetaminophen developed lethargy, hypotension, and respiratory depression.18 Due to the paucity of data, oxycodone is not recommended in patients receiving dialysis.
Codeine: Codeine undergoes biotransformation into many metabolites. Fifty to 70 percent of the parent drug is converted through glucuronidation to codeine-6-glucuronide, 15% to morphine via CYP2D6, and 10% to 15% to norcodeine via CYP3A4, while 5% to 15% is eliminated unchanged in the urine.19 Additionally, codeine’s metabolites are biotransformed further into other metabolites, leading to additional drug accumulation (i.e., M3G and M6G from morphine). In a single-dose study by Guay et al, 60 mg of codeine phosphate was administered to 6 hemodialysis patients and 6 healthy volunteers, resulting in a significantly increased half-life of codeine in the dialysis group when compared to results in the volunteers.20
A case report presented by Talbott et al discussed a pediatric patient with renal failure, who developed respiratory depression from codeine conversion to M6G.21 Such effects may occur in patients who are known as ultra-rapid metabolizers of CYP2D6, converting codeine to morphine and its metabolites much more readily.21 Due to the risk of metabolite accumulation, which has been reported in hemodialysis patients, it recommended to avoid use of codeine in the renal insufficiency or dialysis patient population as well.
Meperidine: Meperidine is metabolized in the liver by hydrolysis to meperidinic acid followed by partial conjugation with glucuronic acid.16 It also undergoes N-demethylation to normeperidine. Normeperidine has an elimination half-life five to 10 times longer than the parent compound.16 Moreover, normeperidine, an active metabolite, is about half as potent as meperidine, but has twice the CNS stimulation effects (i.e., risk of convulsions).22 Renal failure increases the elimination half-life of normeperidine. A case report by Hassan et al describes a patient who received meperidine on continuous cycles of peritoneal dialysis and developed myoclonic contractions and a tonic-clonic seizure.22 Because of the risk of normeperidine accumulation, avoidance in patients with renal impairment and dialysis is recommended.
Fentanyl: Fentanyl is extensively (99%) metabolized via CYP3A4 to norfentanyl, which is an inactive metabolite.23 Fentanyl appears to be safe; however, a concern regarding its clearance reduction in patients with blood urea nitrogen (BUN) >60 mg/dL exists. This reduction in clearance may lead to respiratory depression.23 Exercise caution when dosing this agent; meanwhile, dosage adjust for low GFR (TABLE 4).16
Fentanyl has low water solubility and high protein-binding (80%-85%) properties, rendering it a poor analgesic choice in patients undergoing dialysis.9
Methadone: Methadone is metabolized to pyrrolidine, followed by its conversion to pyrroline.9 Up to 45% of methadone and its metabolite can be eliminated through feces, suggesting that methadone may be used safely in renal disease.24 It is recommended to use caution when dosing methadone in a low GFR population, and to start with lower doses titrating up to effect (TABLE 4).16
A case report by Kreek et al included three patients: an oliguric peritoneal dialysis patient, an anuric hemodialysis patient, and a recent kidney transplant recipient.24 In all three individuals, plasma levels were documented to be in the therapeutic range. Less than 1% of methadone was removed via dialysis24;this was most likely due to the high protein-binding and volume of distribution properties methadone is known to have.9 Therefore, it is recommended to avoid methadone in patients with hemodialysis.
Tramadol: Tramadol is a 4-phenylpiperidine analogue of codeine that has two enantiomers similar in structure to venlafaxine.8,11 The parent compound, mainly a serotonin reuptake inhibitor, is metabolized by CYP2D6 to O-desmethyltramadol, which is a weak mu opioid-receptor agonist.8 The parent compound and metabolite are both 90% excreted in the urine.8 In renal impairment patients or individuals undergoing dialysis, it is recommended to avoid tramadol because of increased risk of toxicity. The increase in toxicity occurs as a result of the decreased clearance of the parent compound and accumulation of the metabolite (TABLE 5).9,16
CONCLUSION
Opioid analgesics may present many challenges to clinicians. It is imperative to carefully monitor chronic pain patients in order to effectively manage and provide optimal pain treatments, while minimizing the potential for adverse effects. Pharmacokinetic and pharmacodynamic properties must be accounted for prior to opioid initiation in patients with hepatic and renal impairment. Extensive clinical data supporting specific dosing recommendations are lacking. More definitive studies are needed to establish guidelines on pain management in organ dysfunction. While the majority of recommendations are currently based on case reports, it is essential to adopt the practice of low-to-high, slow dose titration, until a therapeutic effect is achieved in the hepatic and renal impairment patient population.
REFERENCES
1. Institute of Medicine Report from the Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: The National Academies Press; 2011.
2. Smith H. Opioid metabolism. Mayo Clin Proc. 2009;84(7):613-624.
3. Davidson S, Jhangri G. The impact of chronic pain on depression, sleep, and the desire to withdraw from dialysis in hemodialysis patients. J Pain Symptom Manage. 2005;30(5):465-473.
4. Davison SN. Pain in hemodialysis patients: prevalence, cause, severity, and management. Am J Kidney Dis. 2003;42:1239-1247.
5. Delco F, Tchambaz L, Schlienger R, et al. Dose adjustment in patients with liver disease. Drug Safety. 2005;28(6):529-545.
6. Ishizu H, Shiomi S, Kawamura E, et al. Gastric emptying in patients with chronic liver diseases. Ann Nucl Med. 2002;16:177-182.
7. Verbeeck R. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147-1161.
8. Davis M. Cholestasis and endogenous opioids. Clin Pharmacokinet. 2007;46(10):825-850.
9. Dean M. Opioids in renal failure and dialysis patients. J Pain Symptom Manage. 2004;28:497-504.
10. Smith H, Bruckenthal P. Implications of opioid analgesia for medically complicated patients. Drugs Aging. 2010;27(5):417-433.
11. Trescot A, Datta S, Lee M, et al. Opioid pharmacology. Pain Physician. 2008;11(2 suppl):S133-S153.12. Angst M, Buhrer M, Lotsch J. Insidious intoxication after morphine treatment in renal failure: delayed onset of morphine-6-glucuronide action. Anesthesiology. 2000;92(5):1473-1476.
13. Lee M, Leng M, Tiernan E. Retrospective study of the use of hydromorphone in palliative care patients with normal and abnormal urea and creatinine. Palliat Med. 2001;15(1):26-34.
14. Durnin C, Hind I, Wickens M, et al. Pharmacokinetics of oral immediate-release hydromorphone (Dilaudid IR) in subjects with renal impairment. Proc West Pharmacol Soc. 2001;44:81-82.
15. Kirvela M, Lindgren L, Seppala T. The pharmacokinetics of oxycodone in uremic patients undergoing renal transplantation. J Clin Anesthesia. 1996;8:13-18.16. Johnson SJ. Opioid safety in patients with renal or hepatic dysfunction. Pain Treatment Topics. June 2007. http://paincommunity.org/blog/wp-content/uploads/Opioids-Renal-Hepatic-Dysfunction.pdf. Accessed June 13, 2014.
17. Poyhia R, Seppala T, Olkkola K, et al. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br J Clin Pharmacol. 1992;33:617-621.
18. Foral P, Ineck JR, Nystrom KK. Oxycodone accumulation in a hemodialysis patient. South Med J. 2007;100(2):212-214.
19. Stamer U, Zhang L, Stuber F. Personalized therapy in pain management: where do we stand? Pharmacogenomics. 2010;11(6):843-864.
20. Guay D, Awni W, Dindlay J, et al. Pharmacokinetics and pharmacodynamics of codeine in end-stage renal disease. Clin Pharmacol Ther. 1988;43(1):63-71.
21. Talbott G, Lunn A, Levy F, et al. Respiratory arrest precipitated by codeine in a child with chronic renal failure. Clin Pediatrics. 1997;36(3): 171-173.
22. Hassan H, Bastani B, Gellens M. Successful treatment of norme-peridine neurotoxicity by hemodialysis. Am J Kidney Dis. 2000;35(1): 146-149.
23. Koehntop D, Rodman J. Fentanyl pharmacokinetics in patients undergoing renal transplantation. Pharmacotherapy. 1997;17(4):746-752.
24. Kreek M, Schecter A, Gutjahr C, et al. Methadone use in patients with chronic renal disease. Drug Alcohol Depend. 1980;5(3):197-205.
25. Parab P, Ritschel W, Coyle D, et al. Biopharm Drug Dispos. 1988;9(2): 187-199.
26. Palkama V, Neuvonen P, Olkkola KT, et al. The CYP3A4 inhibitor itraconazole has no effect on the pharmacokinetics of IV fentanyl. Br J Anaesth. 1998;81:598-560.
To comment on this article, contact rdavidson@uspharmacist.com.



