US Pharm. 2010;35:HS-15-HS-19.
A decision by the FDA to approve a drug for market is based on a thorough review of that drug’s safety and efficacy to determine whether the benefits outweigh the risks associated with its use. While clinical trials help to establish the efficacy of a drug and to reveal common adverse events, there are limitations in identifying safety concerns in this setting. For example, study participants may not necessarily represent “real world” patients receiving the drug once it is on the market. Pharmacovigilance involves the continued monitoring of the safety profiles of products throughout their life cycles, and particularly once in the marketplace, through scientific data-gathering activities relating to the detection, assessment, and understanding of adverse events.
Adverse events are undesirable experiences associated with the use of a medical product. In the regulatory setting, adverse events are categorized as adverse drug events (ADEs) and, as a subset of ADEs, adverse drug reactions (ADRs) (TABLE 1).1
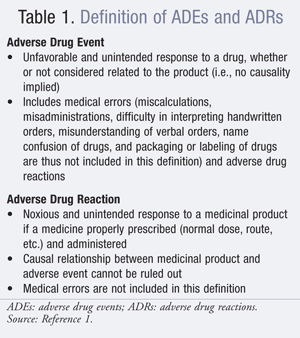
It is challenging to estimate the true incidence of ADEs in the general population, with uncertainty about the number of patients exposed to a given drug, poor documentation, and underreporting, as well as the effect of publicity around a particular drug on the reported frequency of an event. In 2008, the FDA received more than 530,000 reports of suspected ADEs, of which some 33,000 were submitted directly to the FDA; most were submitted by manufacturers.2 That same year, there were 320,000 serious adverse events and nearly 50,000 deaths.3 While reporting of ADEs is a vital component in ensuring drug safety postapproval, the FDA’s Adverse Event Reporting System is believed to capture only a small portion of ADEs that occur.4
While adverse event reporting guidelines are frequently in place at both institutional and professional-society levels, health care professionals (HCPs) are currently not required by federal law or regulation to submit reports of ADEs on any medical product, due to the challenges of enforcing such a mandate.5 Many ADEs are likely never reported because they are not recognized as safety concerns, perhaps because the HCP is unfamiliar with the reporting process and its importance. Moreover, incomplete fields or inaccuracies in the submitted data may limit the utility of the data.
Patients, HCPs, and the pharmaceutical industry all have an interest in enhancing drug safety by reporting suspected ADEs to the FDA. To encourage this, the FDA recently released a final rule entitled, “Toll-Free Number for Reporting Adverse Events on Labeling for Human Drug Products,” requiring the following statement to be distributed with dispensed new and refill prescription drugs: “Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.”6 Moreover, the recent focus on safety concerns surrounding several widely used products has intensified the focus on the drug safety system, and timely and complete ADE reporting continues to be an integral component of this.
The aim of this article is to familiarize the reader with the process for ADE/ADR reporting for drugs currently in the U.S. marketplace. This will include discussion of how the ADE reports are handled by the FDA and the pharmaceutical industry, and the measures currently being used and those under development to monitor patient safety. While there are global ADE reporting requirements, this article will center on ADE reporting to the FDA. Also, for simplicity of reading, the term ADE is used throughout to encompass both ADEs and ADRs.
REPORTING OF ADVERSE EVENTS THROUGH MEDWATCH
MedWatch, established by the FDA in 1993, was designed to expedite and broaden voluntary reports of serious ADEs by HCPs and manufacturers.7 The FDA requests that a serious ADE (i.e., the ADE is fatal, life-threatening, permanently/persistently/significantly disabling, requires initial or prolonged hospitalization, causes congenital anomaly, requires intervention to prevent permanent impairment or damage) be reported if there is suspicion that it is related to the use of one or more medications.7,8 Reporting of a nonserious but unexpected ADE (i.e., not listed in the product information) could also be useful in uncovering previously unidentified adverse effects of drugs. Reports should be submitted even where the HCP completing the report is not certain the product caused the event.
TABLE 2 lists the key information needed when reporting an ADE.9 The importance of completing some of the fields (e.g., description of the event) is obvious. However, the value of information on other fields is often not so apparent. Reporting the lot number, for example, can provide relevant information if there is suspicion of involvement of a counterfeit product, and help with patient follow-up. Including the name of the manufacturer will help identify whether the brand manufacturer’s or the generic company’s product has been administered, to assist with finding the source of ADEs that may be manufacturer specific. All manufacturers, whether a brand or generic company, have the responsibility to appropriately conduct follow-up on ADEs. Currently, however, brand manufacturers are receiving and subsequently reporting to the FDA some ADE reports for products actually dispensed as generic and thus originating from another manufacturer. From January 2004 to December 2007, for example, prescriptions of fluoxetine HCl from Eli Lilly and Company made up only 4.5% of the prescriptions in the U.S., yet Eli Lilly submitted almost 50% of the fluoxetine HCl ADE reports in the publicly available safety database published by the FDA during that time frame (data on file, Eli Lilly and Company).10 Ensuring that each manufacturer appropriately manages ADE reports specific to their own drug is only likely to become more challenging as the number of available generics in the marketplace increases.

While reports should be filed even when all the details are not available, an HCP reporting an ADE should gather as much information as possible when preparing to submit the report so that the FDA and the drug manufacturer have the information required to assess safety and to draw appropriate conclusions.
Channels for Reporting ADEs
ADEs can be reported directly by the HCP or consumer to the FDA using MedWatch, or they can be reported to the manufacturer who in turn reports them to the FDA (FIGURE 1).
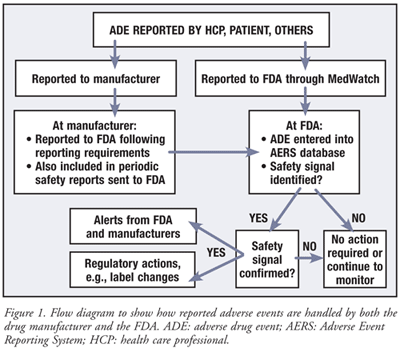
Reporting Directly to the FDA: There are three options for submitting a voluntary report: 1) Complete Form 3500 online at www.accessdata.fda.gov/scripts/medwatch; 2) call 1-800-FDA-1088 to report by telephone; and 3) download a copy of Form 3500 at www.fda.gov/downloads/Safety/MedWatch/DownloadForms/UCM082725.pdf and either fax it to 1-800-FDA-0178 or mail it back (MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787) back using the postage-paid addressed form. This may be most appropriate where there are attachments to submit with the report.
Reporting to the FDA Via the Manufacturer: The majority of ADEs are reported to the drug manufacturer, whose contact information can be found at the relevant manufacturer Web sites. The manufacturer collects all pertinent information from the reporter and is required to send this information to the FDA. Serious and unexpected ADEs are expedited by the manufacturer to the FDA within 15 days of the manufacturer’s receipt of the ADE. All ADEs are also periodically reported by the manufacturer to regulatory agencies through periodic safety reports. These reports contain summaries of all ADE reports in a designated time period and include patient exposure (both postmarketing and clinical trial), presentation of case summaries, study findings, and overall safety evaluation.
WHAT HAPPENS TO ADE REPORTS AT FDA?
At the FDA, ADE reports, including manufacturers’ expedited reports of serious adverse events as well as the manufacturers’ other reports provided within periodic safety updates, are captured in a postmarketing surveillance database, the Adverse Event Reporting System (AERS) (FIGURE 1). The AERS is a computerized database of adverse event reports, containing more than 4 million ADE records.11 Not all ADE reports are entered into the database.11 The FDA screens and assesses all ADE reports received, and prioritizes those entered based upon the associated safety and public health concern.
At the FDA, each ADE case report is evaluated for the adequacy of the information, the temporal association of the product and the event, potentially confounding factors such as patient comorbidities or concomitant drug therapy, and dechallenge-rechallenge information. This helps determine whether the ADE may be related to the use of the drug and whether there may be a safety signal. A safety signal can be defined as a concern about an excess of adverse events compared to what would be predicted to be associated with a product’s use.12 Safety signals may include new adverse events (particularly if serious) that were not seen in clinical trials and thus were not captured on the product label; an apparent increase in the severity of an event seen in clinical trials; occurrence of serious events thought to be extremely rare in the general population; new product-product, product-device, product-food, or product-dietary supplement interactions; identification of a previously unrecognized at-risk population (e.g., populations with specific racial or genetic predispositions or comorbidities); or concerns arising from the way a product is used (e.g., adverse events seen at higher than labeled doses or in populations not recommended for treatment).
It is often not possible to tell from an individual ADE report if there is a causal relationship between the drug and the medical event, and the importance of including as detailed case information as possible when submitting an ADE cannot be overemphasized. In addition to reviewing individual cases, the FDA will use further information provided by the drug manufacturer.
Case reports are assessed at the aggregate level to create a case series to characterize the potential safety risk and to identify risk factors. Creating a case series commonly involves an analysis of the clinical and laboratory manifestations and course of the event; demographic characteristics of patients with events, exposure duration, time from initiation of product exposure to the adverse event; doses used (including labeled doses, greater than labeled doses, and overdoses); use of concomitant medications; the presence of comorbid conditions; and the lot numbers for products used in patients with events. Data mining—utilizing statistical methods to identify potential safety signals from data in the AERS database, as well the large manufacturers’ safety databases—will also help in identifying ADE reporting trends and in directing safety questions to the manufacturer. Other activities to help with risk identification and assessment include the conduct of further trials and observational studies by the manufacturer to assess the risk attributed to drug exposure; creation of registries to collect information from multiple sources (e.g., physician records, hospital summaries, pathology reports); and the conduct of patient and HCP surveys.
OUTCOMES OF ADE REPORTING
If, on completion of review of all available clinical data, the safety signal or signals are confirmed, further steps and actions may be necessary in the interest of patient safety (TABLE 3). Even before these definitive steps are taken, the FDA may at times communicate risk information about the drug safety issues that are under review to the HCP as well as to the general public. This occurs as a result of an early safety signal that suggests an association between the drug and the ADE being evaluated. These actions are designed to assist with decision making around use of these drugs, to prompt discussions between HCP and patient, and to stimulate additional adverse event reporting. Early risk information is communicated through various outlets (i.e., public health advisories, HCP sheets, indexes to drug-specific information, drug safety podcasts, drug safety newsletters, consumer information Web sites), available at the MedWatch and Center for Drug Evaluation and Research (CDER) Web sites of the FDA. An HCP can also sign up to receive safety alerts and information on safety-related label changes by e-mail. If, on clinical data review, a safety signal is not confirmed, the manufacturer and the FDA will continue to monitor relevant safety information.

THE FUTURE OF DRUG SURVEILLANCE
In this article, we have discussed the critical role of ADE reporting in the drug postmarketing safety system. While the resulting AERS data may provide the initial signal of a safety concern that can then be further investigated, there are certain limitations to data in the current system. For example, there is no information on the number of users of a specific drug that can be used to calculate the proportion of patients experiencing an event; there is often inadequate case documentation in the reports; and there is substantial underreporting of ADEs.13
Due to these limitations, the FDA, drug manufacturers, and others (academics, HCPs, professional associations, patient advocacy groups, computer/software companies, managed care organizations, and think tanks) are working together to advance and improve the current system, develop methods for integration of ADE reports with other postmarket safety approaches, and to improve information sharing. In this regard, e-prescriptions, e-health care records, integrated electronic claims, patient information databases, and broad electronic communication targeted to the appropriate health care professionals and patients hold much promise.
While some of these advances may be years away, in a recent draft Prescription Drug User Fee Act (PDUFA) IV Drug Safety Five-Year Plan, the FDA proposes to use the new user fees (fees collected from drug manufacturers to fund the drug review process) to add resources to monitor drug safety of marketed drugs.14 Steps proposed include the adoption of new tools and the improvement of existing tools. Further, under the recently passed FDA Amendments Act of 2007, the FDA will be establishing a postmarket risk identification and analysis system utilizing federal and private sector health-related electronic data.15 Progress to implement the postmarket risk identification system has already occurred. On May 22, 2008, the FDA launched the Sentinel Initiative with the goal of creating and implementing the Sentinel System, an integrated, electronic system for monitoring medical product safety, which will enable the agency to examine existing data sources from the private and public sector, such as electronic health record systems and medical claims databases, for information about medical products. Such a system could improve the FDA’s ability to monitor the performance of a product throughout its life cycle and facilitate data mining and other safety research-related activities.
In the future, HCPs will likely be able to access a patient’s electronic health care record on a wireless computer chart, submit an adverse event report, receive comprehensive information based on the patient’s medical history to communicate immediately to the patient, and order an alternative product all within a matter of seconds. Until then, it is important that the health care community optimize the current tools within the drug safety system, which include timely, complete, and accurate reporting of ADEs. The most updated information on the MedWatch program and ADE reporting can be found by visiting the FDA’s Web site.
CONCLUSION
As a result of recent safety concerns surrounding widely used drugs, there is an increasing focus on drug safety. Timely, complete, and accurate ADE reporting is an essential component of monitoring, and subsequently improving, patient safety.
The authors gratefully acknowledge Patrick DeLisle for his assistance.
REFERENCES
1. Nebeker JR, Barach P, Samore MH. Clarifying adverse drug events: a clinician’s guide to terminology, documentation, and reporting. Ann Intern Med. 2004;140:795-801.
2. US Food and Drug Administration. Reports received and reports entered into AERS by year as of December 31, 2008. www.fda.gov/cder/aers/statistics/aers_received.htm. Accessed April 17, 2009.
3. US Food and Drug Administration. AERS patient outcomes by year as of December 31, 2008. www.fda.gov/cder/aers/statistics/aers_patient_outcome.htm. Accessed March 31, 2009.
4. Drazen JM, Rainey J, Begg H, Butler AS, Rapporteurs. Forum on Drug Discovery, Development and Translation. In: Adverse Drug Event Reporting: The Role of Consumers and Health-Care Professionals. Washington, DC: National Academies Press; 2007:27.
5. Baciu A, Stratton K, Burke SP, eds. Committee on the Assessment of the US Drug Safety System. The Future of Drug Safety: Promoting and Protecting the Health of the Public. Washington, DC: National Academies Press; 2006:chap4:5.
6. US Food and Drug Administration, HHS. Final Rule: Toll-Free Number for Reporting Adverse Events on Labeling for Human Drug Products. Federal Register: October 28, 2008 (vol. 73, Number 209):63886-63897.
7. US Food and Drug Administration. MedWatch Web site. www.fda.gov/medwatch. Accessed March 31, 2009.
8. US Food and Drug Administration. What is a serious adverse event? www.fda.gov/medwatch/report/DESK/advevnt.htm. Accessed March 31, 2009.
9. Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment, FDA 2005.
10. IMS Health. National Prescription Audit (NPA). Plymouth Meeting, PA.
11. US Food and Drug Administration. www.fda.gov/cder/aers/statistics/default.htm. Accessed March 31, 2009.
12. US Food and Drug Administration. Guidance for Industry Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment. March 2005.
13. Committee on the Assessment of the U.S. Drug Safety System, Baciu A, Stratton K, Burke SP, eds. The Future of Drug Safety: Promoting and Protecting the Health of the Public. Washington, DC: National Academies Press; 2006:chap2:20.
14. The FDA Draft PDUFA IV Drug Safety Five-Year Plan, March 2008. www.fda.gov/cder/pdufa/fedreg.htm. Accessed March 31, 2009.
15. The US Food and Drug Administration Amendments Act of 2007 (FDAAA), Pub L No. 110-85.
To comment on this article, contact rdavidson@uspharmacist.com.



