US Pharm. 2012;37(10):HS-12-HS-15.
ABSTRACT: Nerve injuries cause considerable loss of function in many individuals. Therapeutic regimens have been developed to manage the pain associated with nerve injury, as well as to restore the normal functioning of the nerve. Analgesics, anti-inflammatories, antidepressants, and anticonvulsants all provide some degree of pain control. Effective neuroprotective growth factors have been developed, but their use is not without limitations. More recently, the focus has shifted to the neuroprotective pharmacologic factors L-carnitine and N-acetylcysteine, which have exhibited promising results. Surgery to repair the damaged nerve and maximize the number of axons that regenerate through the site of injury may be performed. Despite the availability of these options, there is room for more effective therapies that can restore nerve function.
Various types of trauma can result in nerve injury, a significant cause of functional morbidity.1 Because nerve trauma is the most common form of nervous system trauma encountered in clinical practice, neuronal death due to trauma is an important clinical issue.1 Many drugs used to manage pain due to nerve injury were developed as anticonvulsants and antidepressants, so it is important for pharmacists to be aware of the indication for which the drug is being used, as well as the recommended dosage for the particular indication. Injury grade is an important predictor of possible symptoms, as well as of possible repair.
Mechanisms of Traumatic Nerve Injury
Nerve injury can result from one or more of the following: mechanical trauma; crush injury (such as that seen in fractures, hematomas, compartment syndrome); penetrating traumas, whereby peripheral nerves are partially or completely severed in an irregular manner (stab-wound lacerations, surgical incisions); and stretch injury (nerves can stretch 10%-20% before becoming structurally damaged).2
Grades of Nerve Injury
Classification of nerve injury depends upon the nerve components affected, loss of functionality, and the ability to recover spontaneously.2 Two grading systems are used to stage the extent of nerve injury: Seddon’s system and, more recently, Sunderland’s system.2
Seddon proposed a three-tiered model for nerve injury: neurapraxia, axonotmesis, and neurotmesis, in order of increasing severity. According to this system, the neurapraxial stage involves a reversible conduction block characterized by local ischemia and selective demyelination of the axon sheath.3 The axon’s continuity is retained, and although conduction across the nerve injury is inhibited, conduction within the nerve both proximal and distal to the lesion remains intact.4 The prognosis for an injured nerve at this stage is good, and recovery occurs within weeks to months.4 Wrist drop secondary to prolonged external pressure that compresses the radial nerve at the spiral groove of the humerus is a clinical example of neurapraxia.5
Axonotmesis is a more severe stage of injury, with disruption of not only the myelin sheath, but the axon as well. The epineurium and perineurium remain intact, meaning that there is still some continuity within the nerve.3,4 Axonotmesis leads to Wallerian degeneration, a process whereby the part of the axon that is separated from the neuronal cell body disintegrates distal to the injury.2 The prognosis for nerves at this stage is fair, and recovery may require months.4 Axonotmesis is commonly seen in crush injuries and displaced bone fractures.5
Neurotmesis, the most severe form of nerve injury, is associated with complete nerve division and disruption of the endoneurium.3 In neurotmesis, the axon, myelin sheath, and connective-tissue components are damaged, disrupted, or transected.2 As with axonotmesis, neurotmesis initiates Wallerian degeneration, but the prognosis for nerves is poor. Neurotmesis is commonly seen after lacerations or ischemic injuries.
Sunderland’s classification, which builds upon Seddon’s system,
divides Seddon’s last stage—neurotmesis—into three subcategories.5 There are, therefore, five grades of nerve injury according to Sunderland’s system (TABLE 1).
Nerve Repair and Regeneration
Nerves in the central nervous system do not repair themselves; instead, intact areas take over the function of the damaged areas, a process known as plasticity.6 In contrast, the nerves of the peripheral nervous system (PNS) attempt to regenerate and reinnervate themselves. Peripheral nerves respond to injury or disease in one or more of the following ways: segmental remyelination, Wallerian degeneration, and axonal degeneration.5,6 Segmental demyelination and Wallerian degeneration are repair mechanisms that are relevant to traumatic nerve injury, whereas axonal degeneration is more characteristically seen in metabolic and toxic nerve disorders such as diabetes mellitus and renal failure.5
A number of intrinsic and extrinsic factors are important for successful regeneration of PNS neurons.7 The method of nerve regeneration depends upon the type of injury sustained. Grade I injuries are repaired by remyelination; injuries of more severe grades are repaired by collateral axon sprouting and proximal-to-distal nerve regeneration.5,6 Collateral axonal sprouting is the process by which outgrowths develop from the shafts of existing axons.
Treatment of Nerve Injuries
The treatment of nerve injuries is traced to William A. Hammond, surgeon general of the U.S. Army during the American Civil War.3 The goal of treatment is to restore function to the damaged nerve, improve quality of life, and reduce neuropathic pain.8 It is important to treat not only the nerve, but also exogenous sources of injury.
In severe pain syndrome, which is perhaps the most pressing consideration in early stages, the nerve injury leads to axonal sprouting and neuroma formation.9 Nerve pain, a combination of somatosensory, cognitive, and emotional alterations, is characterized by a burning sensation and dysesthesias.10 Typically neuropathic, this pain is relatively difficult to treat. Severe pain syndrome requires medications that specifically treat neuropathic pain.6 Although a number of options for reducing nerve pain exist, only about 70% of patients are adequately managed; in some patients, combination therapy provides better control.11 A typical strategy is to use short-acting medications that provide relief until longer-acting medications, such as antiepileptics or antidepressants, take effect.5 Refractory pain may require the further addition of an antidepressant or antiepileptic whose mechanism of action (MOA) differs from the MOA of the one already being used. Narcotics are reserved for more aggressive pain control.5
Pharmacologic Therapy
A variety of agents may be used to treat nerve pain (TABLES 2 and 3).
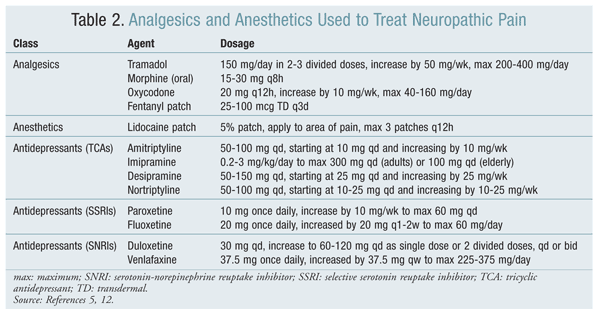

Analgesics: Analgesics provide short-term and immediate control of pain, but the response is generally poor. Analgesics include nonsteroidal anti-inflammatory drugs, tramadol, and opioids.6 Opioid analgesia is dose dependent and related to serum levels, but doses are limited by toxicity and side effects. Anesthetics and topical agents such as capsaicin also provide an analgesic effect; however, capsaicin is inconsistent in its ability to relieve pain and in fact may exacerbate pain, so its use is not recommended.11,12
Antidepressants: Antidepressants—particularly tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors (SNRIs)—are highly effective for reducing pain, but their use is limited by associated side effects.11 TCAs are associated with anticholinergic side effects such as dry mouth, urinary retention, constipation, orthostatic hypotension, and dizziness. Patients taking SSRIs and SNRIs may experience sexual dysfunction and diarrhea in addition to somnolence, dry mouth, and dizziness.
Anticonvulsants: The use of anticonvulsants to manage neuropathic pain has derived from the similarities between the pathophysiological mechanisms of epilepsy and neuropathy.13 It has been observed that changes in the expression of genes encoding the sodium (Na)+ and calcium (Ca)2+ channels lead to alterations in their distribution and composition.9 Changes in the biophysical properties, as well as abnormal accumulation, of Na+ channels in the nociceptors and sensory nerves result in high-frequency ectopic firing in the injured neuron, similar to what occurs with epileptic hyperexcitability.9
Carbamazepine, which is chemically related to TCAs, was the first anticonvulsant studied for the relief of neuropathic pain. Carbamazepine and phenytoin are Na channel blockers that block high-frequency ectopic discharges, an effect that accounts for their protective activity against seizures and neuropathic pain.9 This blocking activity is greater than the Na channel–blocking activity of lidocaine.9 The literature supporting the use of carbamazepine for the management of nerve pain is stronger than that for phenytoin.13
In spinal dorsal horn neurons, wind-up may be generated by Ca-dependent plateau potentials. Wind-up is the process whereby neurons demonstrate a progressive increase in responsiveness with repeated activation of C-fibers. Since antiepileptics such as gabapentin, oxcarbazepine, and lamotrigine target high-voltage activated Ca channels, they are useful in nerve pain management. Gabapentin was originally developed as a gamma-aminobutyric acid (GABA) analogue, although it does not bind to GABA receptors or affect the uptake or metabolism of GABA.13,14 Gabapentin exerts its effect through the alpha-2-delta type of Ca2+ channels, although the exact mechanism is not known. In a number of trials, gabapentin has had the most clearly demonstrated analgesic effect for the treatment of neuropathic pain. Dosing starts at 300 mg per day and may be increased by 300 mg each day until an adequate analgesic response is achieved or the patient experiences dose-limiting side effects.14 Effective dosages range from 900 to 3,600 mg per day in three divided doses.13 Gabapentin is generally well tolerated; the most common side effects are mild-to-moderate dizziness, somnolence, ataxia, and confusion.13,14
Pregabalin has an MOA similar to that of gabapentin, and like gabapentin it does not affect GABA receptors. The analgesic effect of pregabalin can be seen within the first week of treatment, and the effective dosage range is 150 to 600 mg per day orally in two to three divided doses.14 Pregabalin is generally well tolerated, with the most common side effects being dizziness, somnolence, and peripheral edema. The drug is excreted renally, and more than 98% remains unchanged in the urine.
Gabapentin and pregabalin are particularly useful for neuropathic pain in patients with a history of cardiovascular disorders, glaucoma, or urinary retention.11 Lamotrigine blocks sodium channels and consequently inhibits glutamate release. Its potential to modulate and control neuropathic pain is relatively good, although its use is not as well established as that of carbamazepine, gabapentin, or pregabalin. A recent Cochrane review concluded that the antiepileptic lacosamide does not seem to offer any benefits in the reduction of neuropathic pain.15
Nonpharmacologic Therapy
Transcutaneous electrical nerve stimulation may provide pain relief for some patients.11 Antivirals and steroids may be used to decrease endoneurial edema that is associated with peripheral nerve injury in some patients.8
Neuroprotection is the focus of research in the management of nerve injuries. Neuroprotective therapies prevent cell death, and since cell death is a slow process, there is a therapeutic window for neuroprotection.16 The first group of drugs to be studied was the neuroprotective growth factors, but their clinical use was limited for two main reasons: (1) the need for different cocktails of growth factors required for each type of nerve, since the response to different neurotrophic factors varies widely between different neuronal subpopulations, and (2) the limiting side effects associated with these drugs.16,17
More recently, researchers’ interest has shifted to the neuroprotective pharmacologic factors L-carnitine and N-acetylcysteine (NAC). Both have been shown to be safe and effective neuroprotective agents, possibly because of their antioxidant properties.16,17 The signaling of cell death also leads to simultaneous transcription of regenerative genes. The ratio of the genes for cell death to those for regeneration is what ultimately determines the fate of the nerve cell.1 It is thought that both mitochondrial dysregulation and the prevention of entry into the cell cycle result in neuronal death. Since these processes seem to depend upon reactive oxygen species, antioxidants such as L-carnitine and NAC are potentially attractive as neuroprotective agents.
Surgical Treatment
The aim of surgical treatment is to repair the damaged nerve, maximize the number of axons that regenerate through the site of injury, and increase the proportion of axons that grow back to appropriate targets.18 The most appropriate surgical technique that will allow the maximum number of axons to regenerate is based upon the extent of the nerve damage, as well as the nerve’s functional viability and location; the patient’s age and medical conditions; whether functional ability can be restored to some extent with surgery; and whether benefits outweigh risk, costs, and loss of productivity.
Future Developments
To improve pain control in nerve injury, research is focusing not only on the treatment of symptoms, but also on treatment of the cause of the pain. To this end, one option is to focus on imitating the organism’s reaction to limit nerve damage or to enhance the regeneration of injured axons.19 The functional outcome of nerve injury remains inadequate despite advances in surgical techniques.17
REFERENCES
1. Hart AM, Terenghi G, Kellerth JO, Wiberg M. Sensory
neuroprotection, mitochondrial preservation and therapeutic potential of
N-acetyl-cysteine after nerve injury. Neuroscience. 2004;125:91-101.
2. Tuncel U, Turan A, Kostakoglu N. Acute closed radial nerve injury. Asian J Neurosurg. 2011;6:106-109.
3. Browner BD, Jupiter JB, Levine AM, et al. Skeletal Trauma: Basic Science, Management, and Reconstruction. 4th ed. Philadelphia, PA: Saunders; 2009.
4. Nerve injury. Wheeless’ Textbook of Orthopaedics. www.wheelessonline.com/ortho/nerve_injury. Accessed June 15, 2012.
5. Daroff RB, Fenichel GM, Jankovic J, Mazziotta JC. Bradley’s Neurology in Clinical Practice. 6th ed. Philadelphia, PA: Saunders; 2012.
6. Campbell WW. Evaluation and management of peripheral nerve injury. Clin Neurophysiol. 2008;119(9):1951-1965.
7. Bosse F. Extrinsic cellular and molecular mediators of peripheral axonal regeneration. Cell Tissue Res. 2012;349:5-14.
8. Sharon I. Acute nerve injury treatment & management.
http://emedicine.medscape.com/article/249621-treatment. Accessed June
13, 2012.
9. Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685-692.
10. Machelska H. Control of neuropathic pain by immune cells and opioids. CNS Neurol Disord Drug Targets. 2011;10:559-570.
11. Vranken JH. Mechanisms and treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem. 2009;9:71-78.
12. Ferri FF. Ferri’s Clinical Advisor 2013. St. Louis, MO: Mosby; 2012.
13. Tremont-Lukats IW, Megeff C, Backonja MM. Anticonvulsants for neuropathic pain syndromes: mechanisms of action and place in therapy. Drugs. 2000;60:1029-1052.
14. Lynch ME. The pharmacotherapy of chronic pain. Rheum Dis Clin North Am. 2008;34:369-385.
15. Hearn L, Derry S, Moore RA. Lacosamide for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;2:CD009318.
16. Hart AM, Terenghi G, Wiberg M. Neuronal death after peripheral
nerve injury and experimental strategies for neuroprotection. Neurol Res. 2008;30:999-1011.
17. Terenghi G, Hart A, Wiberg M. The nerve injury and the dying neurons: diagnosis and prevention. J Hand Surg Eur Vol. 2011;36:730-734.
18. Fawcett J, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43-60.
19. Martin YB, Herradón G, Ezquerra L. Uncovering new pharmacological
targets to treat neuropathic pain by understanding how the organism
reacts to nerve injury. Curr Pharm Des. 2011;17:434-448.
To comment on this article, contact rdavidson@uspharmacist.com.



