US Pharm. 2011;36(7):HS28-HS32.
Cystic fibrosis (CF) is an autosomal recessive multisystem condition caused by a defect in the gene that encodes the CF transmembrane regulator (CFTR).1 The CFTR is a chloride channel located at the apical surface of epithelial cells. The resulting disruption of CFTR-mediated ion and water transport leads to abnormally viscous secretions that obstruct the lungs, pancreas, biliary tree, intestine, and other organs.2-4
In the lungs specifically, water is reabsorbed as more and more sodium is pulled, causing dehydration and airway surface liquid (ASL) depletion. As a consequence of loss of ciliary stability and function, the mucociliary clearance is reduced. Retention of airway secretions leads to the hallmarks of CF lung disease—chronic bacterial airway infection, an airway inflammatory response, and, finally, irreversible lung damage.3-5 The majority of CF-related morbidity and mortality is a result of chronic pulmonary sepsis; 85% of CF-related deaths are due to pulmonary dysfunction.1,6 With advances in treatment, the life expectancy of a patient with CF has improved in the last decades and at present is 37 years, the improvement being most pronounced in the 2- to 15-year-old group.6-8
Current and Emerging Therapies
Most current therapies attempt to address only the symptomatic complications of the disease, not the underlying problem.3 These include mucolytics that improve mucociliary clearance, drugs that reduce viscoelasticity of the sputum, and antibiotics targeted toward infectious bacteria. In addition, the Cystic Fibrosis Foundation (CFF) approves various physical airway clearance methods such as percussion and postural drainage, autogenic drainage, and high frequency chest wall or airway oscillation devices.6 These techniques are employed to loosen the mucus and assist in its clearance from the lower airways.9
Different research teams have employed varying strategies in order to develop better ways of managing CF. Some of the major therapies will be discussed next and are summarized in TABLE 1.
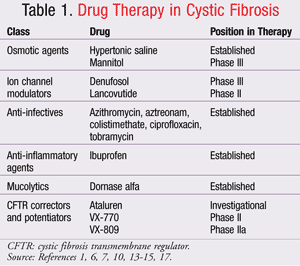
Improving Airway Hydration: Osmotic Agents and Ion Channel Modulators
Osmotic Agents: Nebulized 7% hypertonic saline has been shown to reduce the frequency of pulmonary exacerbations and improve lung function to a lesser extent. It seems to work in three ways: improve mucus hydration, induce coughing, and increase ASL height. It is particularly beneficial in the long term in children with CF who do not yet have significant lung disease.1,3 Some researchers believe it may make the mucus more compact or reduce the mobility of Pseudomonas aeruginosa.6 Currently, it is recommended that patients older than 6 years of age be given nebulized hypertonic saline twice daily.6 For those who cannot tolerate the 7% formulation, strengths as low as 3% may be employed. In some cases, the use of a bronchodilator prior to the saline decreases the incidence of bronchospasm and improves mucus clearance.6
Mannitol is an investigational agent in airway rehydration therapy for CF patients. It is currently undergoing phase III trials as a dry powder formulation.6 While its osmotic action draws water and rehydrates the airways, it is a sugar that can provide biofuel for the growth of bacteria and thereby increase the risk of infection. Mannitol does, however, have a more prolonged effect compared to hypertonic saline since it takes longer to diffuse across epithelial cells.6 Since mannitol induces a cough reflex and may result in bronchospasm, it is advisable to pretreat patients with a bronchodilator. The dry powder formulation of mannitol will be easier to carry and administer than nebulized hypertonic saline.6
Ion Channel Modulators: Further research on ways of improving airway hydration has revealed that chloride secretion in epithelial cells may be increased by stimulating non-CFTR chloride channels. Two molecules, administered via inhalation, are currently under investigation for stimulating calcium-dependent chloride channels in CF airways, namely lancovutide and denufosol.3
Lancovutide is an antibiotic that activates an alternative chloride channel by increasing intracellular calcium.6 Phase II studies have demonstrated a statistically significant increase in the forced expiratory volume in 1 second (FEV1) in patients receiving lancovutide. However, patients commonly reported throat numbness, headache, chest numbness, and gastrointestinal (GI) discomfort.6
Denufosol stimulates the purinergic P2Y2 receptor on epithelial cells, which in turn activates calcium-sensitive chloride channels, allowing chloride to flow into the ASL. Through this pathway, denufosol bypasses the malfunctioning CFTR protein-regulated chloride channel.1,6 Alongside increasing the ASL, denufosol also increases the release of surfactant, mucin, and the mucociliary clearance.6 Denufosol is currently undergoing phase III trials that have so far demonstrated an increased FEV1.6 Adverse effects associated with denufosol include cough, chest tightness, wheezing, and increased sputum.6 It has been postulated that a combination of denufosol and hypertonic saline may be more effective than either of the individual agents on their own, but this hypothesis is yet to be confirmed.6
Anti-Infectives
Anti-infectives used in CF are targeted at reducing chronic pulmonary sepsis, particularly when caused by P aeruginosa. Since infection with P aeruginosa decreases life expectancy and worsens nutritional status, it is vital to prevent this organism from colonizing the airways.10 This can be achieved by initiating therapy early after the onset of a P aeruginosa infection.10
Inhaled antibiotics have been shown to reduce rates of hospitalization, reduce the need for parenteral therapies, and improve weight gain as well as the quality of life.11 More specifically, they decrease the bacterial load, the frequency of respiratory exacerbations requiring supplemental antibiotics, and the decline in pulmonary function.12
Tobramycin 300 mg nebulized twice daily for 28 days has been shown to be effective in early treatment. In cases of chronic colonization, the CFF recommends the continuous use of inhaled tobramycin.10 Tobramycin should not be inhaled <6 hours between regimens. It has a 28-day holiday after treatment. Inhaled colistin, available as colistimethate sodium, is a well-established treatment for P aeruginosa. It is given as a nebulized solution in doses of 30 to 150 mg every 12 to 24 hours. More recently, it has been proposed that the combination of colistin and tobramycin may be more efficient than either of these agents alone.10 It is very likely that dry powder inhalation formulations of antipseudomonal antibacterials, including colistin and tobramycin, will be available on the market soon. Dry powder formulations have the advantage of being more stable and more portable.10
Aztreonam as an inhalation solution was recently approved by the FDA and is used in 75-mg doses delivered three times daily for 28 days followed by a 28-day drug holiday. It has been shown to improve respiratory symptoms and lung function.7 Amikacin, levofloxacin, ciprofloxacin, and a combination of fosfomycin and tobramycin are all soon to be available on the market as inhaled antibiotics.7
Other bacterial species are also recognized in CF, but their role is not fully clear. Methicillin-resistant Staphylococcus aureus (MRSA) is an emerging pathogen in patients with CF and is becoming a growing concern.10 Burkholderia species, a highly virulent group, have innate antibiotic resistance and are transmissible from person to person.13 Patients with CF who acquire Burkholderia cepacia deteriorate rapidly and have higher mortality rates due to the development of cepacia syndrome, a sepsislike syndrome with severe necrotizing pneumonia.5
Not only is azithromycin a well-established oral antibiotic for the long-term management of CF, but it is also thought to play a role as an anti-inflammatory agent, making it one of the drugs of choice for pulmonary infections.1,14 While ciprofloxacin gives good blood concentrations, organisms such as P aeruginosa can quickly develop resistance to it.15
Patients may also develop fungal infections, commonly caused by Candida species and Mycobacterium avium complex (MAC).5 The recommended regimen for the treatment of MAC comprises a macrolide, specifically clarithromycin, ethambutol, or rifampicin. Additionally, an IV aminoglycoside such as streptomycin or amikacin is used in patients who have advanced or previously treated disease.16
Anti-Inflammatory Agents
Anti-inflammatory agents are targeted at managing the persistent inflammatory response that leads to lung damage.1 Inflammatory pathways in CF patients are predominantly signaled by neutrophils. The aim is to establish a balance between the damage caused by chronic inflammation and the protective effects of the host inflammatory defense system.1
In addition to having demonstrated no clinical advantage over placebo, corticosteroids have been associated with adverse effects such as growth retardation, cataract formation, and the development of glucose intolerance.1 This group of anti-inflammatories is therefore no longer used in the management of CF. Presently, ibuprofen is the only anti-inflammatory recommended in CF patients. Due to its established side-effect profile, it is essential that patients on long-term ibuprofen be monitored for GI and renal side effects.14 Inhaled glutathione, phosphodiesterase 5 (PDE-5) inhibitors, oral acetylcysteine HE-3286, simvastatin, methotrexate, docosahexaenoic acid, hydroxychloroquine, pioglitazone, and alpha1-antitrypsin are all under investigation for use as anti-inflammatories in CF.1,7 Since there is no standard procedure for monitoring the extent of inflammation in patients with CF, it is difficult to assess the effectiveness of the agents under investigation.7
Mucolytics
Mucolytic therapy is aimed at facilitating the physiologic clearance of mucus by decreasing its elasticity and viscosity.17 Well established in the treatment of CF is dornase alfa, recombinant human DNase, which enzymatically breaks down the large amounts of DNA in the sputum and reduces sputum viscosity.1,17 It is available as a 2.5-mg/2.5-mL dose given daily via a nebulizer.17 The solution needs to be refrigerated and protected from light. Patients on dornase alfa commonly complain of voice alteration, pharyngitis, laryngitis, rash, chest pain, sore throat, cough, conjunctivitis, and fever. Less commonly, patients may experience GI problems, hypoxia, and weight loss.17
N-acetylcysteine (NAC) breaks disulfide bonds in the mucus, but there is no dependable data demonstrating that it has a clinical benefit in CF so far.1,17 Additionally, it is associated with side effects such as bronchospasm, nausea and vomiting, stomatitis, and rhinorrhea. For these reasons, NAC is not recommended in the management of CF.17
Newer agents that target filamentous actin (F-actin), such as gelsolin and thymosin ß4, are also being developed.17 Actin, released together with DNA from necrosing cells, forms a copolymer with DNA polymers to reduce the viscosity of sputum. Gelsolin, an acting severin protein, and thymosin ß4, a F-actin sequestrant, have both been shown to reduce sputum viscosity and tenacity in this manner.17
CFTR Correctors and Potentiators
Recently, research has been focused on trying to solve the basic problem in CF—the dysfunction of CFTR due to mutations in the gene. While the most common mutations involve a deletion of phenylalanine in position 508, currently over 1,600 mutations have been discovered.3,6 These can be conveniently classified according to the effect they have on the CFTR (TABLE 2).1,3

Currently, CFTR pharmacotherapy has received a lot of attention.1,3 Overall, class I to III mutations are associated with the absence of functional CFTR, while classes IV and V have functional CFTR. The deletion of phenylalanine at position 508 is a class II mutation.1 Agents under investigation are labeled correctors or potentiators.1
The theory behind CFTR correctors is to generate an agent that will correct one or more of the defects found in class II mutations by:
- Rescuing proteins from endoplasmic reticulum (ER) degradation
- Improving trafficking of CFTR to the cell surface
- Inhibiting proteins that are involved in the recycling of CFTR in the cell membrane.3
Several molecules currently under investigation that can function as correctors have been identified using high throughput assays.13 Ataluren (PTC124) is one such investigational molecule that can cause ribosomal read-through of premature stop mutations in patients with class I mutations, correct the processing of CFTR, and thereby increase the production of functional CFTR.1,2 Ataluren lacks both the antibiotic function and toxicity of aminoglycosides, antibiotics that were initially investigated to induce read-through of premature termination codons.3 Another agent labeled VX-809, which has just completed phase IIa trials, acts as a “chaperone” to assist the movement of defective CFTR to the epithelial cell membrane.1,13 Researchers have found that this molecule is well tolerated in CF patients, and further studies are under way.3
CFTR potentiators are called thus because they enhance the activity of CFTR that is correctly located at the cell membrane.13 They would be particularly useful in the rare cases of class III mutations.3 VX-770 is an orally administered drug that creates a more effective opening of the CFTR chloride channel.1 It has demonstrated good efficacy and safety in CF patients as shown by nasal potential difference measurements and is currently undergoing phase II trials.3,9
Soybeans contain an isoflavone known as genistein that improves the response to cyclic adenosine monophosphate (cAMP)-induced chloride secretion in CF patients.18 A similar effect is noted with alkylxanthines and PDE-5 inhibitors.13,18
Ideally, a combination of corrector and potentiator activity might be the ultimate treatment of CF.19 While extensive efforts are being undertaken to discover molecules with such properties, there are very few to date that have demonstrated both characteristics.19
Gene Therapy
Gene replacement is another area of interest for researchers working with the field of CF, particularly the UK Cystic Fibrosis Gene Therapy Consortium. The aim is to deliver a normal CFTR gene to the lung that would result in the expression and restored function of CFTR in the CF airway epithelium.3 While it may seem like a straightforward concept, gene therapy is not without its challenges.13 These include, but are not limited to, determining the best method of delivery and establishing the effective dose.13 The lungs also have innate defense mechanisms that need to be overcome before the target epithelial cells can be successfully reached.9
Lung Transplant
CF patients with end-stage lung disease may opt for a double lung transplant or a heart-lung transplant.20 The prognosis for this, however, is lower than that of other organ transplants, with a 3-year survival for about 60% of patients with CF.20
It is important to note that this review only covers the management of pulmonary complications associated with CF. Treatment of other conditions such as endocrine, bone, and GI disorders caused by CF is beyond the scope of this article.
Conclusion
The aim of new therapies is to treat the basic defect in CFTR in individual patients and hence improve the life expectancy in patients who are born with CFTR mutations. While much research is ongoing in this area, the information gathered so far is not substantial enough to translate into considerable clinical improvements. A multidisciplinary team that includes pharmacists is important in ensuring that the best outcomes are achieved using the tools currently available. Health care professionals involved in the care of patients with CF therefore need to keep abreast with the dynamic changes taking place in the management of this disease.
REFERENCES
1. Jones AM, Helm JM. Emerging treatments in cystic fibrosis. Drugs. 2009;69:2003-2010.
2. Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011 Jan 13; Epub ahead of print.
3. Grasemann H, Ratjen F. Emerging therapies for cystic fibrosis lung disease. Expert Opin Emerg Drugs. 2010;15:653-659.
4. Mogayzel PJ Jr, Flume PA. Update in cystic fibrosis 2009. Am J Respir Crit Care Med. 2010;191:539-544.
5. Strausbaugh SD, Davis PD. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279-288.
6. Pettit RS, Johnson CE. Airway-rehydrating agents for the treatment of cystic fibrosis: past, present, and future. Ann Pharmacother. 2011;45:49-59.
7. Anderson P. Emerging therapies in cystic fibrosis. Ther Adv Respir Dis. 2010;4:187-195.
8. Goss CH, Rosenfeld M. Update on cystic fibrosis epidemiology. Curr Opin Pulm Med. 2004;10:510-514.
9. Kreindler JL. Cystic fibrosis: Exploiting its genetic basis in the hunt for new therapies. Pharmacol Ther. 2010;125:220-229.
10. Cohen-Cymberknoh M, Shoseyov D, Kerem E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;193:1463-1471.
11. Parkins MD, Elborn JS. Aztreonam lysine: a novel inhalational antibiotic for cystic fibrosis. Expert Rev Respir Med. 2010;4:435-444.
12. Doull IJM. Recent advances in cystic fibrosis. Arch Dis Child. 2001;85:62-66.
13. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1991-2004.
14. Narasimhan M, Cohen R. New and investigational treatments in cystic fibrosis Ther Adv Respir Dis. 2011 March 3; Epub ahead of print.
15. Whitehead A. Treating adults with cystic fibrosis. Pharm J. 2001;266:508-510.
16. de Vrankrijker AM, Wolfs TF, van der Ent CK. Challenging and emerging pathogens in cystic fibrosis. Paediatr Respir Rev. 2010;11:246-254.
17. Henke MO, Ratjen F. Mucolytics in cystic fibrosis. Paediatr Respir Rev. 2007;8:24-29.
18. Roomans G. Pharmacological treatment of the ion deficient transport defect in cystic fibrosis. Exp Opin Invest Drugs. 2001;10:1-20.
19. Sheppard DN. Cystic fibrosis: CFTR correctors to the rescue. Chem Biol. 2011;25;19:145-147.
20. Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681-689.
To comment on this article, contact rdavidson@uspharmacist.com.



