US Pharm. 2011;36(10):HS-2-HS-8.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2010–2011 (TABLE 1) detail the basic clinical and pharmacologic profile of each new drug, as well as key precautions and warnings. Also included for each drug is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly shows that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have clearly demonstrated the appearance of “new” adverse reactions for many NMEs within 2 to 3 years of their first becoming available. Some of these drugs may eventually acquire at least one black box warning for serious adverse reactions or are withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature.

Belimumab (Benlysta, Human Genome Sciences, GlaxoSmithKline)
Indication and Clinical Profile1,2: Belimumab has been approved for the treatment of adults with active, autoantibody-positive systemic lupus erythematosus (SLE) who are receiving standard therapy (i.e., corticosteroids, quinoline antimalarials, immunosuppressives, nonsteroidal anti-inflammatory drugs). It is the first in a new class of drugs known as B-lymphocyte stimulator (BLyS)–specific inhibitors. Prior to belimumab, the last drugs approved by the FDA to treat lupus were hydroxychloroquine (Plaquenil), aspirin, and corticosteroids.
Two clinical studies involving 1,684 patients with lupus demonstrated the safety and effectiveness of belimumab. Patients with active lupus were randomly assigned to belimumab plus standard therapy or an inactive infused solution (placebo) plus standard therapy. Patients with prior B cell–targeted therapy or IV cyclophosphamide and those with active lupus involving the kidneys or central nervous system (CNS) were excluded. Patients given belimumab plus standard therapy experienced less disease activity than those given placebo plus standard therapy. Results suggested that certain patients had a reduced likelihood of severe flares, and some patients were able to reduce their steroid doses. African American patients appeared not to respond to belimumab treatment. To address this finding, an additional study will be conducted to further evaluate the safety and effectiveness of belimumab in this subgroup of lupus patients. The efficacy of belimumab has not been evaluated in patients with severe active lupus nephritis or severe active CNS lupus or in combination with other biologics or IV cyclophosphamide. Thus, belimumab use is not recommended in these situations.
Pharmacology and Pharmacokinetics1,2: BLyS is a naturally occurring protein that was discovered in 1996. Belimumab is a human immunoglobulin G (IgG) 1-gamma monoclonal antibody that specifically blocks the binding of soluble BLyS—a B-cell survival factor—to its receptors on B cells. It does not bind B cells directly, but by binding BLyS belimumab inhibits the survival of B cells, including autoreactive B cells, and reduces the differentiation of B cells into immunoglobulin-producing plasma cells.
Overall, belimumab’s pharmacokinetics appears to be linear across the typical dosage range. Following IV administration, serum belimumab concentrations decline in a biexponential manner, with a mean distribution-phase half-life of 1.0 to 2.2 days and a mean terminal elimination half-life of 8.5 to 14.1 days. Mean clearance after a single IV dose is approximately 7 mL/day/kg—much less than the glomerular filtration rate, indicating that renal clearance is not a major component of belimumab clearance. Concomitant use of immunosuppressants, hydroxychloroquine, or prednisone during the study had no significant effects on belimumab pharmacokinetics. No formal studies were conducted to examine the effects of renal or hepatic impairment on the pharmacokinetics of belimumab; thus, dose modification does not appear to be necessary.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions in clinical studies were nausea, diarrhea, and fever. Patients also commonly experienced infusion reactions, so pretreatment with an antihistamine should be considered. In clinical studies, more deaths and serious infections were reported in belimumab patients compared with placebo patients. Belimumab should not be administered with live vaccines. The manufacturer is required to provide a patient medication guide advising of the risks associated with belimumab. Belimumab is a Pregnancy Category C drug and should not be used in nursing women.
Concurrent use of other SLE therapies (mycophenolate, azathioprine, methotrexate, antimalarials, nonsteroidal anti-inflammatory drugs, aspirin) and hydroxymethyl glutaryl coenzyme A reductase inhibitors does not appear to significantly influence belimumab pharmacokinetics. Coadministration of steroids and ACE inhibitors may result in an increase in belimumab clearance, but this does not appear to be clinically significant. The effect of belimumab on the pharmacokinetics of other drugs has not been evaluated.
Dosage and Administration1,2: Belimumab is available as a lyophilized powder in single-use vials (120 mg/vial and 400 mg/vial) for IV infusion only; it must be reconstituted prior to administration. The recommended dosage is 10 mg/kg at 2-week intervals for the first three doses and at 4-week intervals thereafter. Antihistamine premedication for prophylaxis against infusion reactions and hypersensitivity reactions should be considered prior to dosing with belimumab. If the patient develops an infusion reaction during administration, the infusion rate may be slowed or interrupted. Infusion should be discontinued immediately if the patient experiences a serious hypersensitivity reaction.
Ceftaroline (Teflaro, Forest Pharmaceuticals)
Indication and Clinical Profile3,4: Ceftaroline fosamil, a broad-spectrum bactericidal cephalosporin, was approved for the treatment of community-acquired bacterial pneumonia (CABP) (including cases caused by Streptococcus pneumoniae bacteremia) and acute bacterial skin and skin-structure infection (ABSSSI) (including cases caused by methicillin-resistant Staphylococcus aureus [MRSA]). In 2007, pneumonia was the eighth leading cause of death in the U.S. and the primary cause of death from infectious diseases. S pneumoniae accounts for 60% to 70% of all bacterial CABP cases, and pneumococcal strains have an intermediate resistance rate of 28% and a high resistance rate of 16% to current therapies. ABSSSIs, which are among the most common infections treated in the hospital setting, are caused by both gram-positive and gram-negative pathogens. MRSA is a group of Staphylococcus strains that are resistant to many antimicrobial drug classes, including all of the previously approved beta-lactams. In the U.S., MRSA is the most frequent cause of ABSSSI cases presenting to emergency departments and was the reported cause of more than 18,000 deaths in 2005. In the community, most MRSA infections involve the skin. Severe or potentially life-threatening MRSA infections occur most frequently in patients exposed to health care settings.
The safety and efficacy of ceftaroline fosamil were evaluated in four phase III clinical trials in patients aged >18 years. In the two CABP trials, the comparator antibacterial treatment was ceftriaxone (Rocephin); in the ABSSSI trials, the comparator antibacterial treatment was vancomycin (Vancocin) plus aztreonam (Azactam). In the CABP trials, 1,231 patients received ceftaroline or ceftriaxone. Clinical response—improvement in signs and symptoms of pneumonia on day 4 after therapy initiation—served as the key analysis endpoint. In both trials, the effectiveness of ceftaroline was comparable to that of ceftriaxone. In the two ABSSSI trials, 1,396 patients received ceftaroline or vancomycin plus aztreonam. Clinical response—cessation of lesion spread and absence of fever on day 3—served as the key analysis endpoint. In both trials, ceftaroline was comparable to vancomyin plus aztreonam.
Pharmacology and Pharmacokinetics3,4: Ceftaroline (FIGURE 1) is a broad-spectrum cephalosporin with bactericidal activity against both gram-positive and gram-negative pathogens. Like other beta-lactams, its bactericidal action is mediated through binding to essential penicillin-binding proteins (PBPs), which prevent repair and construction of the bacterial cell wall. Ceftaroline is effective against susceptible pathogens commonly associated with ABSSSI, among them S aureus (including methicillin-susceptible and methicillin-resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Escherichia coli, Klebsiella pneumoniae, and Klebsiella oxytoca. It also is effective against susceptible gram-positive and gram-negative bacteria associated with CABP: S pneumoniae (including cases with concurrent bacteremia), S aureus (methicillin-susceptible isolates only), Haemophilus influenzae, K pneumoniae, K oxytoca, and E coli. Ceftaroline is bactericidal against S aureus owing to its affinity for PBP2a, and against S pneumoniae owing to its affinity for PBP2x.
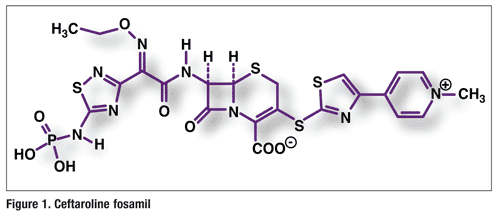
Post IV administration, ceftaroline fosamil is hydrolyzed by plasma phosphatases to its active form, ceftaroline. Further hydrolysis of the ceftaroline beta-lactam ring results in drug inactivation. Ceftaroline and its metabolites are eliminated primarily by the kidneys via glomerular filtration. Based on this clearance profile, dosage adjustment is recommended in patients with moderate-to-severe renal impairment. Since ceftaroline does not undergo significant hepatic metabolism, its clearance is not expected to be significantly altered by hepatic impairment.
Adverse Reactions and Drug Interactions3,4: In clinical trials, the most common adverse reactions occurring in >2% of patients receiving ceftaroline were diarrhea, nausea, and rash. As with other beta-lactams, hypersensitivity reactions including anaphylaxis and anaphylactoid reactions have been reported with ceftaroline. Thus, before ceftaroline therapy is instituted, careful inquiry about previous hypersensitivity reactions to other beta-lactams should be made. If an allergic reaction to ceftaroline occurs, the drug should be discontinued and emergency treatment initiated. Clostridium difficile–associated diarrhea has been reported for nearly all antibacterial agents, including ceftaroline, and may range in severity from mild diarrhea to fatal colitis. Ceftaroline should be used in pregnant or nursing women only if the potential benefit outweighs the potential risk to the fetus or child (Pregnancy Category B).
No clinical drug–drug interaction studies have been conducted with ceftaroline. In vitro studies in human liver microsomes indicated that neither ceftaroline fosamil nor ceftaroline inhibits the major CYP450 isoenzymes. Therefore, neither ceftaroline fosamil nor ceftaroline is expected to inhibit or induce the clearance of drugs metabolized by these metabolic pathways in a clinically relevant manner.
Dosage and Administration3,4: Ceftaroline is supplied as 600 mg or 400 mg of sterile powder for reconstitution in a solution for IV administration. The recommended dosage for both indications, in patients aged >18 years, is 600 mg administered every 12 hours by IV infusion over 1 hour. Dosage adjustment is necessary per manufacturer’s recommendations in patients with moderate (>30- <50 mL/min) or severe (<30 mL/min) renal impairment. The duration of therapy should be guided by the site and severity of infection and by the patient’s clinical and bacteriologic progress.
Denosumab (Xgeva, Amgen)
Indication and Clinical Profile5,6: Denosumab is the first and only RANK ligand (RANKL) inhibitor approved for the prevention of skeletal-related events (SREs) in patients with bone metastases from solid tumors. Bone metastasis occurs in more than 1.5 million cancer patients worldwide and is most commonly associated with cancers of the prostate, lung, and breast, with incidence rates as high as 75% in patients with metastatic disease. SREs include fractures, spinal cord compression, and severe bone pain that may require surgery or radiation. Such events can profoundly disrupt a patient’s life and cause disability and pain. The total economic burden of patients with bone metastases in the U.S. is estimated to be $12.6 billion annually.
Denosumab was approved following a 6-month FDA priority review, a designation reserved for drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists. Approval was based on results from three pivotal, phase III, head-to-head trials evaluating denosumab administered every 4 weeks as a 120-mg subcutaneous (SC) injection versus zoledronic acid (Zometa) given every 4 weeks via a 15-minute IV infusion and adjusted for renal function. The clinical program spanned more than 50 tumor types in more than 5,700 patients. Denosumab demonstrated a clinically significant improvement in SREs compared with zoledronic acid. Specifically, in patients with breast or prostate cancer and bone metastases, denosumab was superior to zoledronic acid in reducing the risk of SREs. In patients with bone metastasis due to other solid tumors or bone lesions due to multiple myeloma, denosumab was noninferior to zoledronic acid. Currently, denosumab is not indicated for the prevention of SREs in patients with multiple myeloma.
Pharmacology and Pharmacokinetics5,6: Denosumab is a fully human IgG2 monoclonal antibody that binds to RANKL, a transmembrane or soluble protein essential for the formation, function, and survival of osteoclasts, the cells responsible for bone resorption. Thus, denosumab prevents RANKL from activating its receptor—RANK—on the surface of osteoclasts and their precursors. Prevention of the RANKL/RANK interaction inhibits osteoclast formation, function, and survival, thereby decreasing bone resorption and increasing bone mass and strength in both cortical and trabecular bone.
Following SC administration, denosumab’s bioavailability was 62%. With multiple SC doses at the recommended dosage interval, up to a 2.8-fold accumulation in serum denosumab concentrations was observed, and steady state was achieved by 6 months. The mean elimination half-life of denosumab is 28 days, and the drug’s pharmacokinetics does not appear to be affected by age, gender, race, or renal or hepatic impairment.
Adverse Reactions and Drug Interactions5,6: In clinical trials, the overall rates of adverse reactions and serious adverse reactions were generally similar between denosumab and zoledronic acid. The most common adverse reactions in patients receiving denosumab were fatigue/asthenia, hypophosphatemia, and nausea, and the most common serious adverse reaction was dyspnea. Osteonecrosis and hypocalcemia were the most common adverse reactions resulting in drug discontinuation. Osteonecrosis of the jaw was infrequent, with no statistically significant difference between treatment arms. Hypocalcemia was more frequent in the denosumab arm. Denosumab is a Pregnancy Category C drug, and it is not known whether the drug is excreted in human milk. Thus, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
No formal drug–drug interaction trials have been conducted with denosumab. In clinical trials, there was no evidence that various anticancer treatments affected denosumab’s efficacy or pharmacokinetics.
Dosage and Administration5,6: Denosumab is supplied as a 120 mg/1.7 mL (70 mg/mL) solution in a single-use vial. The recommended dosage of denosumab is 120 mg administered as an SC injection every 4 weeks in the upper arm, the upper thigh, or the abdomen. Calcium and vitamin D should be administered to the patient as necessary to treat or prevent hypocalcemia.
Eribulin (Halaven, Eisai)
Indication and Clinical Profile7,8: Eribulin is indicated for the treatment of patients with metastatic breast cancer who previously underwent at least two chemotherapeutic regimens for metastatic disease. Breast cancer is the second leading cause of cancer-related death in women. This year, an estimated 207,090 women will be diagnosed with breast cancer, and 39,840 women will die from it. Other therapies used to treat late-stage, refractory breast cancer include Xeloda (capecitabine), for patients with breast cancer resistant to paclitaxel and anthracycline-containing chemotherapy; Ixempra (ixabepilone), for patients with late-stage disease after failure of an anthracycline, taxane, and Xeloda; and Ixempra plus Xeloda, for patients with late-stage disease after failure of anthracycline-based and taxane-based chemotherapy.
The approval of eribulin was based on a controlled multicenter trial involving 762 subjects with metastatic breast cancer who received at least two chemotherapeutic regimens and experienced disease progression within 6 months of their last chemotherapeutic regimen. Subjects were randomized to receive eribulin or a single-agent therapy selected prior to randomization. Randomization was stratified by geographic region, human epidermal growth factor receptor 2 status, and prior capecitabine exposure. Eribulin was dosed at 1.4 mg/m2 on days 1 and 8 of a 21-day cycle, with a median of five cycles of therapy. Treatment in the control arm consisted of 97% chemotherapy and 3% hormone therapy. The main efficacy outcome was overall survival. Median overall survival in subjects receiving eribulin was 13.1 months, compared with 10.6 months in those who received single-agent therapy.
Pharmacology and Pharmacokinetics7,8: Eribulin mesylate is a nontaxane microtubule dynamics inhibitor (FIGURE 2). This agent is a synthetic analogue of halichondrin B, a product isolated from the marine sponge Halichondria okadai. Eribulin exerts its effects via a tubulin-based antimitotic mechanism that inhibits the growth phase of microtubules without affecting the shortening phase and sequesters tubulin into nonproductive aggregates. This leads to G2/M cell-cycle block, the disruption of mitotic spindles, and, ultimately, apoptotic cell death after prolonged mitotic blockage.
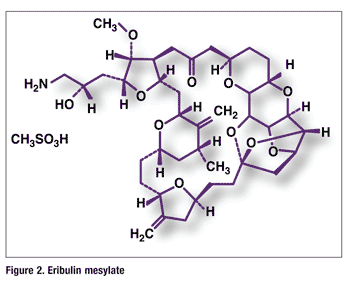
The pharmacokinetics of eribulin is linear, with a mean elimination half-life of approximately 40 hours and plasma protein binding ranging from 49% to 65%. Eribulin exposure after multiple dosing is comparable to that following a single dose. No accumulation of eribulin is observed with weekly administration. Unchanged eribulin was the major circulating species in plasma, with metabolites accounting for <0.6% of the parent compound. There is minimal metabolism by CYP3A4. Eribulin is a weak inhibitor of CYP3A4, but it produces no significant inhibition or induction of other clinically relevant cytochrome isozymes.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions ( >25%) reported with eribulin were neutropenia, anemia, asthenia/fatigue, alopecia, peripheral neuropathy, nausea, and constipation. The most common serious adverse reactions were febrile neutropenia (4%) and neutropenia (2%). The most common adverse reaction resulting in discontinuation of eribulin was peripheral neuropathy (5%). Eribulin has been reported to prolong the QT interval; thus, it should be avoided in patients with congenital long QT syndrome. ECG monitoring is recommended when therapy is initiated in patients with congestive heart failure, bradyarrhythmias, or electrolyte abnormalities and in those taking drugs known to prolong the QT interval (Class Ia and III antiarrhythmics). Hypokalemia or hypomagnesemia should be corrected prior to therapy initiation, and electrolytes should be monitored periodically during therapy. Eribulin is a microtubule inhibitor; therefore, it is expected to cause fetal harm when administered to a pregnant woman (Pregnancy Category D).
No drug–drug interactions are expected with CYP3A4 inhibitors or P-glycoprotein inhibitors. Eribulin does not inhibit CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 enzymes or induce CYP1A2, CYP2C9, CYP2C19, or CYP3A4 enzymes at relevant clinical concentrations. Thus, eribulin is not expected to alter the plasma concentrations of drugs that are substrates of these enzymes.
Dosage and Administration7,8: Eribulin mesylate is supplied as a 1 mg/2 mL (0.5 mg/mL) solution for IV injection. The recommended dosage is 1.4 mg/m2 administered IV over 2 to 5 minutes on days 1 and 8 of a 21-day cycle. The dosage should be reduced to 1.1 mg/m2 in patients with mild hepatic impairment or moderate renal impairment (on the same cycle), and to 0.7 mg/m2 in patients with moderate hepatic impairment (Child-Pugh Class B). Additional modifications, including dose delays or reductions, may be required based on the severity of adverse events such as peripheral neuropathy or reductions in neutrophil or platelet counts (see manufacturer’s literature).
Tesamorelin (Egrifta, EMD Serono)
Indication and Clinical Profile9,10: The FDA approved tesamorelin, a synthetic analogue of growth hormone-releasing factor (GHRF), as the first and only treatment for reducing excess abdominal fat in HIV-infected patients with lipodystrophy (abdominal lipohypertrophy). Several factors, including the antiretroviral drug regimen and the HIV virus itself, are thought to contribute to HIV-associated lipodystrophy, which is characterized by body-composition changes that may include accumulation of excess abdominal fat (abdominal lipohypertrophy).
The efficacy and safety of tesamorelin were determined in two multicenter, randomized, double-blind, placebo-controlled, phase III trials (consisting of a 26-week main phase and a 26-week extension phase) involving 816 HIV-infected patients with excess abdominal fat associated with lipodystrophy. These trials demonstrated statistically significant reductions in visceral adipose tissue and waist circumference versus placebo in HIV-infected patients with excess abdominal fat associated with lipodystrophy. Furthermore, the reductions in visceral adipose tissue and waist circumference observed after 26 weeks of treatment were sustained in patients who received tesamorelin for 52 weeks. Since the long-term cardiovascular safety and potential benefits of tesamorelin treatment have not been demonstrated, careful consideration should be given to whether to continue tesamorelin therapy in patients not showing a clear efficacy response. Data do not indicate improved compliance with antiretroviral therapies in HIV-positive patients taking tesamorelin. Tesamorelin is not indicated for weight-loss management.
Pharmacology and Pharmacokinetics9,10: Tesamorelin (FIGURE 3) is a synthetic analogue of GHRF, a hypothalamic peptide that acts on the pituitary cells in the brain to stimulate the synthesis and release of endogenous GH and serum insulin-like growth factor 1 (IGF-1). GH and IGF-1 exert their effects by interacting with specific receptors on a variety of target cells, including chondrocytes, osteoblasts, myocytes, hepatocytes, and adipocytes, resulting in a host of pharmacodynamic effects. Tesamorelin binds and stimulates human GHRF receptors with similar potency as the endogenous GHRF. Through this mechanism tesamorelin promotes GH release, which reduces visceral fat in HIV-infected patients with excess abdominal fat associated with lipodystrophy. No clinically significant changes in levels of other pituitary hormones, including thyroid-stimulating hormone, luteinizing hormone, adrenocorticotropic hormone, and prolactin, were noted with tesamorelin.
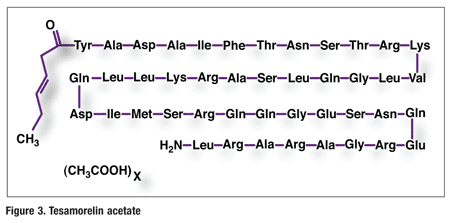
The absolute bioavailability of tesamorelin after SC administration of a 2-mg dose is <4%, and the mean volume of distribution is approximately 10 L/kg. The drug’s metabolism has not been studied but does not appear to involve cytochrome enzymes, as is true for most protein drugs. The mean elimination half-life of tesamorelin is about 25 to 40 minutes after SC administration for 14 consecutive days.
Adverse Reactions and Drug Interactions9,10: The most commonly reported adverse reactions (>5% and more frequent than placebo) involve injection-site reactions (erythema and pruritus) and arthralgia, myalgia, and peripheral edema. In the event of more serious hypersensitivity reactions (e.g., urticaria), tesamorelin should be discontinued immediately and the patient treated promptly. Edema, arthralgias, and myalgias appear to be related to induction of GH and IGF-1 secretion by the drug and are transient or resolve upon discontinuation of treatment. These hormones may promote glucose intolerance, so all patients taking tesamorelin should be monitored periodically for changes in glucose metabolism to identify impaired glucose tolerance or diabetes.
Tesamorelin is contraindicated in pregnancy (Category X) because its action could alter the normal visceral adipose tissue increases associated with pregnancy and possibly cause fetal harm. Tesamorelin also has been associated with birth defects in animal studies. Because of the potential for HIV-1 transmission and serious adverse reactions in nursing infants, women receiving tesamorelin should be instructed not to breastfeed. Other contraindications include disruption of the hypothalamic-pituitary axis due to hypophysectomy, hypopituitarism, pituitary tumor/surgery, head irradiation, or head trauma and known hypersensitivity to tesamorelin and/or mannitol (excipient). Tesamorelin is contraindicated in patients with active malignancies, either newly diagnosed or recurrent. Any preexisting malignancy should be inactive and its treatment completed prior to institution of tesamorelin therapy.
Tesamorelin has no significant effect on CYP3A activity, but its ability to interact with other cytochrome isozymes has not been studied. GH has been reported to modulate cytochrome isozymes involved in corticosteroid, sex steroid, anticonvulsant, and cyclosporine metabolism. Therefore, since tesamorelin stimulates GH production, careful monitoring is advised when it is administered in combination with other drugs known to be metabolized by CYP450 liver enzymes.
Dosage and Administration9,10: Tesamorelin is supplied in a vial containing 1 mg of the drug as a lyophilized powder. The diluent (Sterile Water for Injection, 10 mL) is provided in a separate vial. The recommended dosage is 2 mg injected SC once daily in the abdomen. To reduce the incidence of injection-site reactions, the injection site should be rotated to different areas of the abdomen. Tesamorelin should not be injected into scar tissue, bruises, or the navel. Safety, efficacy, and pharmacokinetics in patients with renal or hepatic impairment have not been established.
REFERENCES
1. Benlysta (belimumab) product information. Rockville, MD: Human Genome Sciences, Inc, and Research Triangle Park, NC: GlaxoSmithKline; March 2011.
2. Navarra S, Guzman R, Gallacher A, et al. Belimumab, a BLyS-specific inhibitor, reduced disease activity, flares and prednisone use in patients with active SLE: efficacy and safety results from the Phase 3 BLISS-52 study. Paper presented at: 73rd Annual Scientific Meeting of the American College of Rheumatology; October 17-21, 2009; Philadelphia, PA.
3. Teflaro (ceftaroline fosamil) product information. St. Louis, MO: Forest Pharmaceuticals, Inc; April 2011.
4. File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis. 2010;51:1395-1405.
5. Xgeva (denosumab) product information. Thousand Oaks, CA: Amgen; November 2010.
6. Lewiecki EM, Miller PD, McClung MR, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22:1832-1841.
7. Halaven (eribulin mesylate) product information. Woodcliff Lake, NJ: Eisai Inc; November 2010.
8. Twelves C, Cortes J, Vahdat LT, et al. Phase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic breast cancer. Clin Breast Cancer. 2010;10:160-163.
9. Egrifta (tesamorelin acetate) product information. Rockland, MA: EMD Serono, Inc; November 2010.
10. Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357:2359-2370.
To comment on this article, contact rdavidson@uspharmacist.com.



