US Pharm. 2012;37(7):38-41.
Sarcoidosis, an inflammatory disease that often affects various systems, is characterized by the presence of noncaseating granulomas caused by the accumulation of inflammatory cells (FIGURE 1).1-3 The diagnosis of sarcoidosis one of by exclusion because of the nonspecific nature of granulomas, which may occur in other disease states, such as certain infections and malignancies.1 To make the diagnosis, involvement of two or more organs is required.1 Sarcoidosis often affects the liver, skin, and eyes, but the lung is the most commonly affected organ.1
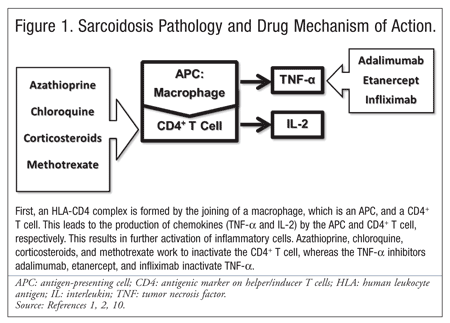
While a definitive cause for sarcoidosis has not been established, it is theorized that exposure to an infectious or noninfectious environmental agent stimulates an inflammatory response in susceptible individuals.1 It has been proposed that the immune response, involving antigen-presenting cells, T cells, and cytokine and chemokine release, stimulates cell recruitment that results in granuloma formation in affected tissues.4 Pathogens that may be responsible for the granulomatous response include Propionibacterium acnes, Borrelia burgdorferi, Mycobacterium tuberculosis and other mycobacteria, Mycoplasma species, and viruses such as herpes.5,6
Sarcoidosis has been observed throughout the world, with Nordic countries having the highest prevalence.1 In the United States, African American individuals are 3.5 times more likely than white individuals to develop sarcoidosis.7 Sarcoidosis is commonly diagnosed in adults who are otherwise deemed healthy.1 Females have a slightly higher risk than males, and health care workers and individuals exposed to insecticides and mold are at higher risk for the disease.1
Clinical Presentation and Diagnosis
Up to one-third of patients with sarcoidosis are asymptomatic.1 Because the lung is the most commonly affected organ, the most common presenting symptoms are of respiratory origin and include cough and dyspnea.1 Many patients present with a symptom history spanning several weeks.1 Moreover, as a result of nonspecific pulmonary symptoms, patients may cycle through the health care system for up to a year before a diagnosis is reached.1
Chest x-ray is standard practice for detecting lung disease. There are five radiographic stages of pulmonary changes, ranging from 0 to IV.1,5 Stage 0 is characterized by a normal chest radiograph, stage I involves bilateral hilar adenopathy, and stage II includes both bilateral adenopathy and the presence of pulmonary infiltrates.5 Stage III involves the presence of pulmonary infiltrates, and stage IV is characterized by pulmonary fibrosis.5
The American Thoracic Society (ATS) recommends that the initial patient workup provide histologic confirmation of the disease, delineate the extent and severity of organ involvement, assess disease stability, and determine whether therapy is indicated.5 Additionally, the ATS has set forth a stepwise approach for further evaluation of a patient with sarcoidosis once the diagnosis has been confirmed.5 The patient history should be comprehensive, including symptoms as well as documentation of occupational and/or environmental exposure to infectious agents or environmental triggers, such as mold.5 Besides a physical examination, chest radiography and pulmonary function testing (spirometry and diffusing capacity of lung for carbon monoxide) should be performed.5 Routine blood and urine chemistries should be collected.5 Additionally, the patient should undergo an ECG, an ophthalmologic examination, and tuberculin skin testing.5
In some instances, a CT scan of the lungs is indicated. According to the ATS, indications include atypical clinical presentation and/or chest radiographic findings, complications of lung disease (e.g., malignancy), and clinical implication of disease with normal chest radiographic findings.5 Common CT findings associated with sarcoidosis are bronchiolar nodules (widespread small nodules with bronchovascular and subpleural distribution), thickened interlobular septae, pulmonary architectural distortion, and conglomerate masses.5
Airway involvement in sarcoidosis can be extensive, ranging from the nasal and oral passages to deep into bronchiolar tissue (TABLE 1).8 Common findings include mucosal nodules and bronchiectases.8 Additionally, airway hyperreactivity and distortion result in airflow limitation.8 In the initial stages of sarcoidosis, airway inflammation gives way to symptoms such as mucosal edema and erythema, as well as to the formation of granulomas.8 As the disease progresses, the mucosa may become granular, cobblestone-like, and nodular.8 Later, a fibrotic phase commences in which tissue scarring may result in narrowed airways.8 It should be noted that pulmonary function may or may not decline with structural airway abnormalities.8 In fact, pulmonary function testing may be within normal limits despite severe structural changes confirmed by imaging studies.8

Treatment
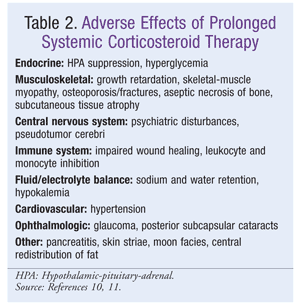
An estimated 30% to 70% of patients diagnosed with sarcoidosis do not require therapy.9 However, in patients with ongoing symptomatic pulmonary sarcoidosis, systemic therapy may be necessary (FIGURE 1).1-3,10 Moreover, asymptomatic patients experiencing a progressive decline in lung function or persistent pulmonary infiltrates also may require therapy.5 The mainstay of treatment is systemic corticosteroid therapy, which—because of the chronic nature of the disease—has a long duration and may result in various adverse effects (TABLE 2).10,11 Alternative steroid-sparing treatment options include cytotoxic agents, antimalarial agents, and tumor necrosis factor (TNF) inhibitors.5,12
Corticosteroids: Because of a lack of definitive criteria, corticosteroid therapy in pulmonary sarcoidosis is initiated on an individual basis.13,14 Systemic corticosteroid treatment may be needed at various times in the disease cycle from disease onset through follow-up, depending upon the individual circumstances.9 Likewise, the dosage and duration of treatment with oral corticosteroids must be individualized, and corticosteroids should be tapered when discontinuation is deemed necessary.5 Because of the possibility of severe adverse effects with long-term systemic corticosteroid therapy, close observation may preclude treatment in patients who are asymptomatic or who have mild pulmonary dysfunction.13 If pulmonary dysfunction progresses to a mild-to-moderate state, observation should be continued for 3 to 6 months, after which treatment may be warranted in cases of pulmonary deterioration and increased disease severity.13
Generally, the starting dose of prednisone or an equivalent is 20 to 40 mg per day on alternate days.5 It is recommended that treatment continue for at least 12 months, with therapeutic evaluation taking place 1 to 3 months after treatment initiation.5 Depending upon disease severity, a patient may respond adequately to a 3-month or 6-month course.5 A lack of response after 3 months of treatment warrants evaluation of other factors, such as noncompliance, corticosteroid resistance, and fibrotic disease.5 Proper follow-up is necessary to monitor for relapse in patients completing a course of therapy.5 If relapse occurs, long-term low-dose therapy may be required.5
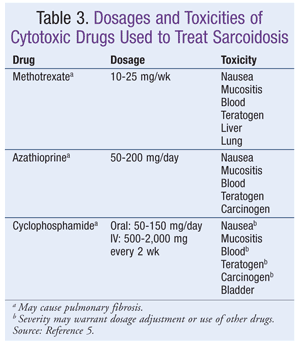
Cytotoxic Agents: Cytotoxic drugs have been used as an alternative treatment in sarcoidosis, but there is no clear rule about when to initiate therapy.5 Among these agents, methotrexate and azathioprine are preferred, with cyclophosphamide held in reserve for refractory cases.5 Methotrexate and azathioprine have demonstrated similar efficacy in the treatment of chronic sarcoidosis.5 Chlorambucil used in combination with low-dose prednisone has achieved response rates similar to those of methotrexate and azathioprine, although the risk of malignancy associated with chlorambucil has limited its use in this disease.5 Similarly, cyclophosphamide has been used in refractory cases, but its use is restricted by its toxicities.5 Common toxicities associated with the use of cytotoxic agents include nausea, mucositis, hematologic abnormalities, teratogenicity, and organ damage.5 TABLE 3 lists the respective dosages and toxicities of various cytotoxic agents used to treat sarcoidosis.5 Pulmonary fibrosis is a known potential adverse effect of all of these drugs.15
Other Agents: Chloroquine has been shown to be beneficial, particularly in patients with lupus pernio and hypercalcemia.5 However, because of the risks of retinopathy and blindness, chloroquine use beyond 6 months is not recommended.5 Hydroxychloroquine is preferred over chloroquine because of the lower risk of ocular toxicity.5
TNF inhibitors, including etanercept, infliximab, and adalimumab, have also been utilized in the treatment of sarcoidosis.12 Certain factors are associated with improved response to TNF-inhibitor therapy, including lower vital capacity, significant dyspnea, impaired quality of life, longer disease duration, and refractory extrapulmonary disease.12 Factors warranting further study include elevated C-reactive protein values and TNF-308 genotype GG.12 A phase II randomized, controlled trial found improved lung function with infliximab dosed at 3 mg/kg or 5 mg/kg at weeks 0, 2, 6, 12, 18, and 24 in addition to corticosteroid and/or immunosuppressive therapy.16 Nonresponse to TNF-inhibitor therapy should prompt an investigation into complications, such as infection or antibody formation.12 Nonresponders may show improvement with alternative dosing regimens or the addition of a cytotoxic agent.12
Lung transplantation has been successful in advanced cases of sarcoidosis.5,17 It has been shown that sarcoid granulomas that derive from the recipient may recur in the donor organ, although patients may not show clinical symptoms of disease.17,18 While lung transplantation remains a lifesaving option for patients with end-stage sarcoidosis, close clinical, radiographic, and histologic monitoring is required to evaluate the efficacy of the transplant.17
Treatment of Complications
In addition to inflammation, infection and other complications may manifest (TABLE 4).5 Aspergillomas are a common complication of sarcoidosis, especially in the more advanced stages of disease, in which fibrosis is present.19 Fatal hemoptysis is the second most common cause of death in sarcoidosis, and close monitoring is vital in patients who develop an aspergilloma so that medical and/or surgical treatment can be initiated if hemoptysis occurs.19,20 Regular chest x-rays are recommended in cases of cystic disease in order to monitor for aspergilloma development.19 Additionally, serum screening for precipitins to Aspergillus antigens should be performed in patients at risk for aspergilloma.19 Itraconazole has been used to treat aspergillomas, although its chronic use may lead to resistance.21 In such cases, voriconazole may be an alternative.20

Osteoporosis secondary to long-term corticosteroid therapy is a common complication of sarcoidosis.5 Common strategies for osteoporosis prevention, such as calcium and vitamin D supplementation, nasal calcitonin, and bisphosphonates, may be utilized in patients with sarcoidosis.5 It is important to note that sarcoidosis may cause hypercalciuria and hypercalcemia; therefore, patients should be monitored while receiving calcium supplementation therapy in order to ensure proper calcium homeostasis.5
Patients with sarcoidosis should be evaluated for fatigue and myalgia.5 Pulmonary rehabilitation may be considered in patients who present with these symptoms, as well as respiratory insufficiency.5 Supplemental oxygen therapy may be indicated in cases of hypoxemia.5
Conclusion
Sarcoidosis is an inflammatory disease that often affects otherwise healthy young adults. The disease occurs worldwide, but a definitive cause has not been established. Exposure to certain environmental and infectious agents is believed to cause sarcoidosis, and it is theorized that the immune response to such agents results in the formation of the classic finding of granulomatous tissue. The absence of symptoms or the presence of nonspecific pulmonary symptoms upon presentation renders diagnosis challenging. Airway involvement may be extensive, and treatment often requires long-term administration of systemic corticosteroids. The continual use of corticosteroids can cause treatment-related conditions such as osteoporosis, as well as abnormalities in several other organ systems. In addition to treatment-related complications, sarcoidosis can result in other complications, such as pulmonary fungal infection. Because of the progressive nature of the disease, it is important to properly screen for suspected sarcoidosis, as extensive pulmonary dysfunction can lead to the need for a lung transplant in severe cases.
REFERENCES
1. Baughman RP, Lower EE. Sarcoidosis. In: Longo DL, Fauci A, Kasper D, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Medical; 2011.
2. Johnson HJ, Schonder KS. Solid-organ transplantation. In: DiPiro J, Talbert RL, Yee G, eds. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill Medical; 2011:1537-1558.
3. Hall PD, Weimert Pilch N, Atchley DH. Function and evaluation of the immune system. In: DiPiro J, Talbert RL, Yee G, eds. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw Hill Medical; 2011:1487-1503.
4. Iannuzzi MC. Advances in the genetics of sarcoidosis. Proc Am Thorac Soc. 2007;4:457-460.
5. Statement on sarcoidosis. Joint Statement of the American Thoracic
Society (ATS), the European Respiratory Society (ERS), and the World
Association of Sarcoidosis and Other Granulomatous Disorders (WASOG)
adopted by the ATS Board of Directors and by the ERS Executive
Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736-755.
6. Drake WP, Pei Z, Pride DT, et al. Molecular analysis of sarcoidosis tissues for Mycobacterium species DNA. Emerg Infect Dis. 2002;8:1334-1341.
7. Rybicki BA, Major M, Popovich J Jr, et al. Racial differences in
sarcoidosis incidence: a 5-year study in a health mainenance
organization. Am J Epidemiol. 1997;145:234-241.
8. Polychronopoulos VS, Prakash UB. Airway involvement in sarcoidosis. Chest. 2009;136:1371-1380.
9. Nunes H, Bouvry D, Soler P, Valeyre D. Sarcoidosis. Orphanet J Rare Dis. 2007;2:46-53.
10. Morgenthau AS, Iannuzzi MC. Recent advances in sarcoidosis. Chest. 2011;139:174-182.
11. Kelly HW, Sorkness CA. Asthma. In: DiPiro J, Talbert RL, Yee G, eds. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw Hill Medical; 2011:439-469.
12. Baughman RP, Lower EE, Drent M. Inhibitors of tumor necrosis factor (TNF) in sarcoidosis: who, what, and how to use them. Sarcoidosis Vasc Diffuse Lung Dis. 2008;25:76-89.
13. Judson MA. An approach to the treatment of pulmonary sarcoidosis with corticosteroids: the six phases of treatment. Chest. 1999;115:1158-1165.
14. Coker RK. Management strategies for pulmonary sarcoidosis. Ther Clin Risk Manag. 2009;5:575-584.
15. Raissy HH, Harkins M, Marshik PL. Drug-induced pulmonary diseases. In: DiPiro J, Talbert RL, Yee G, eds. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill Medical; 2011:511-523.
16. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174:795-802.
17. Martinez FJ, Orens JB, Deeb M, et al. Recurrence of sarcoidosis following bilateral allogeneic lung transplantation. Chest. 1994;106:1597-1599.
18. Milman N, Andersen CB, Burton CM, Iversen M. Recurrent sarcoid
granulomas in a transplanted lung derive from recipient immune cells. Eur Respir J. 2005;26:549-552.
19. Wollschlager C, Khan F. Aspergillomas complicating sarcoidosis. A prospective study in 100 patients. Chest. 1984;86:585-588.
20. Israel HL, Lenchner GS, Atkinson GW. Sarcoidosis and aspergilloma. The role of surgery. Chest. 1982;82:430-432.
21. Dannaoui E, Garcia-Hermoso D, Naccache JM, et al. Use of
voriconazole in a patient with aspergilloma caused by an
itraconazole-resistant strain of Aspergillus fumigatus. J Med Microbiol. 2006;55:1457-1459.
To comment on this article, contact rdavidson@uspharmacist.com.



