US Pharm. 2011;36(6):HS12-HS16.
Fanconi syndrome is a disease that is associated with dysfunction of the proximal tubule of the kidney. It is characterized by the wasting of phosphate, amino acids, glucose, and bicarbonate in varying degrees.1 Clinical manifestations stem from direct or indirect disturbances of the tubule system.2 Fanconi in children and adult populations frequently presents with bone diseases as sequelae of the syndrome.
Emil Abderhalden first investigated Fanconi syndrome in 1903. Finding cysteine crystals in an infant child, he termed it “a familial cystine diathesis.”3 Guido Fanconi, a Swiss pediatrician, later gave his contributions to the understanding of the disease when he noted similarities in cases he had encountered. Subjects in these cases presented with dwarfism, hypophosphatemia, and organic acid excretion into the urine.3
Advances in uncovering the mechanisms responsible for this disease continue to elude investigators. Pharmacists in an institutional setting can have a profound impact by familiarizing themselves with the treatment options available for patients who present with Fanconi syndrome.
Epidemiology and Pathophysiology
Fanconi syndrome is a rare disease with sporadic incidence and reporting of newly diagnosed cases.4 Fanconi syndrome may be caused by inherited, acquired, or exogenous factors (TABLE 1).5 Its morbidity is secondary to the metabolic abnormalities it generates.3 For instance, phosphaturia, glycosuria, and renal tubular acidosis are abnormalities that may develop as a result of the disease. These can affect the development of bone growth, which could stimulate the manifestations of bone disease. In studies reviewed, individuals at risk are most often young Caucasian children.3 For other forms of Fanconi syndrome, both acquired and exogenous, there are no significant populations adversely affected. The age of those affected varies because the etiology is diverse. If the disease was acquired from medications, metal toxicity, or exposure to other noxious agents, it can present itself at any age. However, if Fanconi syndrome was inherited in an autosomal recessive pattern, the onset is usually evident in early development.3

Proteins and solutes are reabsorbed by the proximal tubules using specialized transporters and channels (FIGURE 1). These are localized in the tubular cell membranes, located on the luminal or basolateral membrane.5 The tubules are also responsible for the regulation of acid-base balance, mineral homeostasis, and drug elimination. In Fanconi syndrome, the solutes are prevented from crossing the apical network of the proximal renal tubule cell.5 Patients have significant biochemical and transport carrier abnormalities that result in the wasting of significant proteins, electrolytes, and other solutes. Amino acids, glucose, phosphate, and bicarbonate are transported by these carriers. There is currently no well-defined model or mechanism that highlights understanding of the pathophysiology. However, recent evidence supports the popular theory that the multiplicity of defective transporters accounts for the wasting of solutes. It is far more plausible that the sum of the metabolic disorders reduces the availability of adenosine triphosphate (ATP) for the enzyme Na/K ATPase.6 This reduces the gradient for solute transport resulting in severe solute dysfunction.
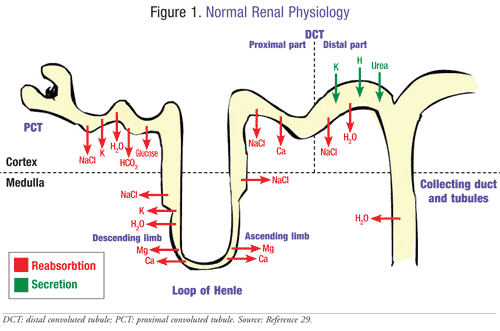
Cystinosis is the most common genetic cause of Fanconi syndrome due to the defective function of cystinosin resulting from the mutation of the gene CTNS, which leads to intralysosomal cystine accumulation.7 This then leads to early onset of the infantile type of cystinosis, which is characterized by severe renal proximal tubular dysfunction during the first year of development. Cystinosin functions as a proton-driven transporter responsible for the export of cystine from lysosomes.8 Adequate evidence is lacking for the link between intralysosomal cystine accumulation and Fanconi syndrome.7 Hereditary Fanconi syndrome is usually accompanied by cystinosis, though the condition can also accompany other transmitted inherited metabolic diseases such as Wilson’s disease and Lowe syndrome.8
Drugs that induce mitochondrial dysfunction have the potential to cause Fanconi syndrome.9,10 The most common drugs are outdated tetracycline antibiotics, chemotherapy agents, antiviral drugs, aminoglycosides, and anticonvulsants. These medications may induce Fanconi syndrome by a variety of different mechanisms. Tetracycline metabolites can cause renal tubular disease with electrolyte imbalance and induce tubular damage within 2 to 8 days after beginning treatment. Reversal of Fanconi syndrome can take up to a year.9 Recent trials with animals suggested that ifosfamide, cisplatin, and carboplatin decrease renal blood flow caused by vascular resistance.9 Cisplatin is a direct toxin to the proximal tubular cells, resulting in an increase in b2-microglobulin and/or aminoaciduria and/or proteinuria.9
Tenofovir and adefovir induce Fanconi syndrome in AIDS/HIV patients.10-12 AIDS/HIV does not directly lead to Fanconi syndrome; rather, it is the antiviral drug metabolites that are responsible for drug-induced Fanconi syndrome.13 Histologically, HIV-induced nephropathy is characterized by the apoptosis of tubular cells. Clinical and experimental evidence has suggested that renal injury may be due to the virus infection because the proximal tubular cells express HIV-specific receptors and coreceptors.9 The mitochondrial toxicity has been established; however, the number of patients using tenofovir is specifically low.14
Nephrotoxicity is a consequence of aminoglycoside administration in hospitalized patients.15 Although amino-glycoside-induced Fanconi syndrome is rare, health care professionals should carefully monitor the use of these antibiotics in critically ill patients; monitoring is specifically more important when managing fluids, electrolytes, and nutrition. Aminoglycosides are reabsorbed in the proximal tubule, causing a decreased glomerular filtration rate. The pathophysiology is uncertain, but it is believed that aminoglycosides irreversibly bind to the cellular membranes causing lysosomal swelling.15,16
Fanconi syndrome represents the extreme end of the spectrum of valproic acid–induced renal impairment.17 Previous studies have shown that the renal impairment stems from the epilepsy itself, not the drug. Additionally, valproic acid–induced Fanconi syndrome appears to be exclusive to children and usually abates on the discontinuation of the drug.17
Clinical Presentation
Clinical presentations of Fanconi syndrome are due to various defects in proximal tubular transport. These include impaired reabsorption of glucose, phosphate, amino acids, bicarbonate, uric acid, water, potassium, and sodium.18 Hereditary Fanconi syndrome features proximal tubular renal acidosis, hypophosphatemic rickets, hypokalemia, polyuria, and polydipsia (usually in infancy).18 Fanconi is the result of cystinosis, growth retardation, depigmentation of the retina, interstitial nephritis, and progressive renal failure. Acquired Fanconi syndrome presents with slightly different abnormalities such as renal tubular acidosis, hypophosphatemia, hypokalemia, osteomalacia, and muscle weakness (TABLE 2).18

Treatment
The primary therapy for Fanconi syndrome is to treat the underlying causes and replace substances wasted in the urine. Fluids and electrolytes are administered via oral or parenteral routes due to dehydration resulting from polyuria, which may exceed 2 to 6 liters per day of dilute urine in cystinosis patients.19 In an acute setting, the patient may require large bolus volumes to initially restore total body water and should be monitored frequently. The decision for oral or parental rehydration depends on the ability of the patient to take oral fluids, the total estimated volume needed to restore losses, and the perceived severity of dehydration.
Metabolic acidosis is caused by the impaired capacity of the kidney to absorb normal levels of bicarbonate. In an acute setting, small boluses of IV sodium bicarbonate may be utilized to raise blood pH.20 For chronic therapy, bicarbonate is administered orally ranging from 3 to 15 mEq/kg/day; however, it has a bitter taste and may cause gastrointestinal (GI) upset.20-24 Citrate salts are dosed based on the amount of bicarbonate equivalents they generate. For example, Cytra-K, which contains potassium citrate and citric acid, generates 2 mEq of potassium and 2 mEq of bicarbonate per mL.25 Adding bicarbonate to the blood may cause cells to increase potassium uptake; therefore, monitoring of serum electrolytes is imperative in patients already predisposed to hypokalemia due to Fanconi syndrome. In order to minimize volume expansion and excretion of bicarbonate, a weak diuretic such as hydrochlorothiazide is utilized at a dose of 1 to 3 mg/kg/day. However, this diuretic is not potassium sparing; therefore, a potassium supplement should be prescribed.
Metabolic acidosis should be corrected by the bicarbonate replacement fluid, but it alone will not correct the pathogenesis of bone disease. Due to the significant loss of phosphate, its supplementation, along with vitamin D, is also necessary. Optimal serum phosphate levels can be achieved with 1 to 3 g/day of supplemental phosphate. The patient should be started on the lowest dose, then titrated up to optimal ranges to avoid GI irritation. For prophylaxis against hypokalemia, 20 mEq orally given daily can be started and then titrated as needed up to 100 mEq given five times a day for adults.26,27 Children may receive 3 to 8 mEq/kg/day orally in divided doses one to five times daily.27 Vitamin D supplementation should be administered in its active form due to possible impaired liver function.
Underlying genetic causes usually involve a defective enzyme in nutrient metabolism resulting in damage to the proximal tubule. Patients with hereditary galactosemia, fructose intolerance, and tyrosinemia would benefit from limiting the amount of galactose, fructose, and tyrosine or phenylalanine, respectively, from their diets.21 Patients with Wilson’s disease should be encouraged to ingest a low-copper diet (such as restricting organ meats, shellfish, and whole wheat) and take D-penicillamine due to studies suggesting beneficial outcomes.4,28 Exercise should also be limited since organ failure or significant decrease of muscle strength could result from genetic conditions.
Cysteamine (Cystagon) is an FDA-approved medication used to treat cystinosis.27,28 It functions by reacting with and then exporting the cystine trapped within lysosomes, thus reducing the load available to induce cellular damage. Initial doses are one-fourth to one-sixth of the maintenance dose and should be titrated up to the initial maintenance dose over 4 to 6 weeks. For pediatric patients, the initial maintenance dose is 1.3 g/m2/day divided four times per day. For patients over 12 years old and greater than 110 lb, the initial maintenance dose is 500 mg four times per day. The goal of therapy is to keep leukocyte cystine levels below 1 nmol of half-cystine/mg protein when measured 5 to 6 hours after drug administration. The doses for both age groups may be titrated to a maximum dose of 1.95 g/m2/day to reach this goal. Side effects include lethargy, diarrhea, seizures, GI toxicity, anemia, and tremor. There are no known drug interactions with cysteamine. Patients allergic to penicillamine should be carefully monitored for adverse events when given cysteamine.27
Monitoring
Monitoring patients with Fanconi syndrome is a crucial part of therapy regardless of the pathogenesis or etiology of the disease (TABLE 3). Internal and external factors affecting Fanconi syndrome are frequency of patient visits, degree of severity, fluid and electrolyte balance, and the efficacy and side effects of medications.5 Patients with acquired Fanconi syndrome may be managed by preventing exposure to toxins (e.g., lead, outdated tetracyclines, aminoglycosides) and alleviating symptoms in children with dietary modifications. Patient prognosis is dependent upon the cause of the syndrome and the severity of renal and extrarenal manifestations. Genetic forms are difficult to manage, are usually associated with disruptions in growth, and involve other organs.5

Parents of young children should be counseled on strategies to prevent or decrease symptoms of the disease. Advising parents to prevent their children from exposure to lead and avoiding outdated antibiotics is an important part of child care. Children with Fanconi syndrome that is secondary to galactosemia or tyrosinemia should be given special dietary instructions to eliminate intolerable nutrients from their diet.5
Conclusion
The extensive clinical presentations of Fanconi syndrome create numerous difficulties for health care professionals. Effectively diagnosing and treating patients is a concern because of the limited information available regarding the pathophysiology of the disease. Although the treatment of underlying complications and electrolyte imbalances has sufficient options, therapeutic advances still need to be made. Further research involving this rare disease is necessary to evolve current practice. It is vital that pharmacists be knowledgeable about the role that medications play in both the cause and treatment of Fanconi syndrome. Understanding the available pharmacologic treatments can help improve patient symptoms and overall outcomes.
REFERENCES
1. Fanconi syndrome. In: Kaplan J, Beers M, Berkwits MMH, eds, et al. Merck Manual of Diagnosis and Therapy. 18th ed. Whitehouse Station, NJ: Merck Research Laboratories; 2006:1079.
2. Baum M. The cellular basis of Fanconi syndrome. Hosp Pract (Off Ed). 1993;28:137-142.
3. Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Brenner BM, ed. Brenner & Rector’s The Kidney. Vol 2. 7th ed. Philadelphia, PA: Saunders; 2004:1697-1707.
4. Brewer G. Practical recommendations and new therapies for Wilson disease. Drugs. 1995;50:240-249.
5. Fathallah-Shaykh S, Spitzer A. Fanconi syndrome. eMedicine. Updated June 30, 2008. http://emedicine.medscape.com/
6. Foreman J. Cystinosis and Fanconi syndromes. In: Barrett T, Avner E, Harmon W, eds. Pediatric Nephrology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:593-607.
7. Foreman JJ, Roth CK. Human renal Fanconi syndrome—then and now. Nephron. 1989;51:301-306.
8. Garces S, Mendonca V, Vaz R, et al. Idiopathic Fanconi syndrome, epilepsy, and liver cirrhosis: is there a link? Kidney. 2009;18:121-124.
9. Harrison HE. The Fanconi syndrome: review. J Chronic Dis. 1958;7:346-355.
10. Izzedine H, Launay-Vacher V, Isnard-Bagnis C, Deray G. Drug-induced Fanconi’s syndrome. Am J Kidney Dis. 2003;41:292-309.
11. Van’T Hoff WG. Molecular developments in renal tubulopathies. Arch Dis Child. 2000;83:189-191.
12. Gupta S. Tenofovir-associated Fanconi syndrome: review of the FDA adverse event reporting system. AIDS Patient Care STDs. 2008;22:99-103.
13. Earle K, Seneviratne T, Shaker J, Shoback D. Fanconi’s syndrome in HIV+ adults: report of three cases and literature review. J Bone Min Res. 2004;19:714-721.
14. James C, Steinhaus M, Szabo S, Dressler R. Tenofovir-related nephrotoxicity: case report and review of the literature. Pharmacotherapy. 2004;24:415-418.
15. Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940-5949.
16. Melnick J, Baum M, Thompson J. Aminoglycoside-induced Fanconi’s syndrome. Am J Kidney Dis.
17. Zaki E, Springate J. Renal injury from valproic acid: case report and literature review. Pediatr Neurol.
18. Laube G, Haq M, Hoff W. Exfoliated human proximal tubular cells: a model of cystinosis and Fanconi syndrome. Pediatr Nephrol. 2005;20:136.
19. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111-121.
20. Quinn A, Sinert R. Metabolic acidosis in emergency medicine: treatment and medication. eMedicine. Updated November 13, 2009. http://emedicine.medscape.com/ 1994;23:188-122. 2002;27:318-319.
21. Rudolph AM, Rudolph CD, eds. Rudolph’s Pediatrics. 21st ed. New York, NY: McGraw-Hill Medical Publishing Division; 2003. STAT!Ref Online Electronic Medical Library. http://online.statref.com/
22. Stein JH, et al, ed. Internal Medicine. Mosby, Inc; 1998. STAT!Ref Online Electronic Medical Library. http://online.statref.com/
23. Hay WW Jr, Levin MJ, Sondheimer JM, Deterding RR, eds. Current Diagnosis & Treatment in Pediatrics. New York, NY: Lange Medical Books/McGraw-Hill; 2009. STAT!Ref Online Electronic Medical Library. http://online.statref.com/
24. American College of Physicians. ACP PIER & AHFS DI Essentials. Philadelphia, PA: American College of Physicians; 2010. STAT!Ref Online Electronic Medical Library. http://online.statref.com/
25. Facts & Comparisons 4.0 [Internet database]. Indianapolis, IN: Wolters Kluwer Health, Inc; 2009. http://online.
26. Cohn JN, Kowey P, Whelton PK, Prisant M. New guideline for potassium replacement in clinical practice. Arch Intern Med. 2000;160:2429-2436.
27. Micromedex Healthcare Series [Internet database]. Greenwood Village, CO: Thomson Healthcare. www.micromedex.com. Accessed March 16, 2011.
28. Schneider J, Clark K, Greene A, et al. Recent advances in the treatment of cystinosis. J Inherit Metab Dis. 1995;18:387-397.
29. Renal physiology and diuretics. Medicopedia. November 25, 2005. http://medicopedia.blogspot.
To comment on this article, contact rdavidson@uspharmacist.com.



