US Pharm. 2010;35(5)(Oncology suppl):13-17.
ABSTRACT: Myelodysplastic syndromes (MDS), a heterogeneous group of clonal bone-marrow disorders, are characterized by ineffective hematopoiesis resulting in blood cytopenias and an increased risk of transformation to acute myeloid leukemia (AML). Until recently, supportive care was the mainstay of treatment for MDS. Within the past decade, three agents have been approved by the FDA for the treatment of MDS: lenalidomide (Revlimid), azacitidine (Vidaza), and decitabine (Dacogen). Despite advances in treatment, complete remission rates and overall survival remain relatively low.
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal bone-marrow disorders characterized by ineffective hematopoiesis resulting in blood cytopenias (anemia, neutropenia, and thrombocytopenia). In addition, MDS often may progress to acute myeloid leukemia (AML).1 MDS predominately affect the elderly, with a majority of patients over the age of 70 years, and have an incidence of more than 10,000 new diagnoses per year in the United States.2,3 Although the incidence rate of MDS has increased over the past decade, this is attributed to an improved awareness and understanding of the disease, as well as to the aging of the population. Patients with MDS suffer the effects of cytopenias, including fatigue, cardiac problems, iron overload, infections, and bleeding complications.1
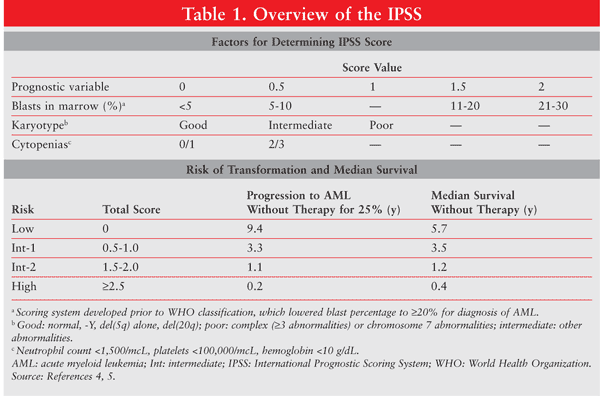
MDS historically have been classified by the French-American-British (FAB) classification system, and more recently by the World Health Organization (WHO) classification.1 Unlike the case with other hematologic malignancies, prognostic models have been developed to predict the risk of AML transformation and overall survival (OS) in MDS patients. The most widely used prognostic system is the International Prognostic Scoring System (IPSS), which identifies four risk classes based on cytopenias, cytogenetics (karyotype), and bone-marrow blasts (TABLE 1) and aids in treatment decisions.4-6
PRINCIPLES OF THERAPY
In formulating a treatment plan for patients with MDS, many patient and disease characteristics are important to consider. These include age, performance status, comorbidities, donor availability, WHO or FAB classification, IPSS score, and cytogenetics. Treatment decisions typically differ between lower-risk disease (IPSS Low or Intermediate [Int]-1) and higher-risk disease (IPSS Int-2 or High). The goals of therapy for lower-risk patients are to relieve transfusion burden, improve quality of life, and restore normal bone-marrow function with low-intensity regimens. The goals of therapy for higher-risk patients are to induce complete remission, prolong survival, and improve quality of life using hypomethylating agents and high-intensity treatments.1,6
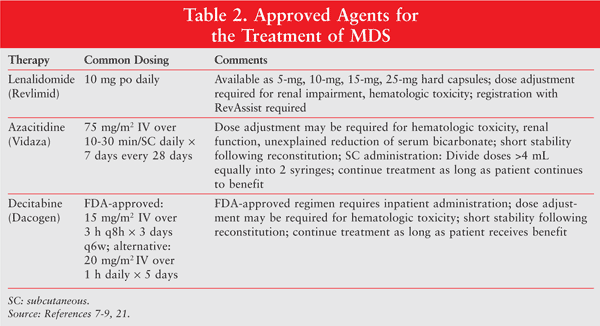
For many years, supportive care was the main treatment option for patients with MDS. Supportive care includes blood and platelet transfusions, growth factors for anemia and neutropenia, and antimicrobials for infection. In the last decade, three agents for the treatment of MDS have been approved by the FDA: the immunomodulating agent lenalidomide and the hypomethylating agents azacitidine and decitabine (TABLE 2).7-9 Unfortunately, despite advances in the treatment of MDS, allogeneic stem cell transplantation remains the only potential cure, and its use is often limited owing to the older age and comorbidities of most patients. Clinical trials remain important treatment options for all patients with MDS.
SUPPORTIVE CARE
Erythropoiesis-stimulating agents (ESAs), with or without granulocyte- or granulocyte-macrophage colony-stimulating factors, are acceptable therapeutic options for initial therapy in lower-risk patients with symptomatic anemia.6 To maximize the use of growth factors, response can be predicted based on transfusion requirements and serum erythropoietin (EPO) levels. Patients with low transfusion needs (<2 units of packed red blood cells [pRBC]/month) and serum EPO levels <500 IU are predicted to have a good response to growth factors, whereas those with high transfusion needs ( >2 units of pRBC/month) and serum EPO >500 IU are predicted to have a poor response.10 Owing to negative data concerning the use of ESAs in solid tumors, ESAs should be discontinued when the hemoglobin is >12 g/dL.
Iron overload, a consequence of cumulative blood transfusions, may lead to increased morbidity and mortality in MDS patients. Multiple guidelines recommend iron chelation in patients with lower-risk disease who require chronic transfusions; patients who have received 20 to 40 units of pRBC; and patients in whom long-term transfusion therapy, as well as survival, is likely.10 Although deferoxamine has traditionally been used for iron chelation, it has a cumbersome route of administration. Deferasirox, an oral iron chelator, was approved in 2005 for the treatment of chronic iron overload due to blood transfusions; several studies evaluating its efficacy in MDS have been published or are ongoing. Unfortunately, iron chelation is expensive and, to date, has not shown a clear survival benefit in the MDS population.10
NEW THERAPIES
Immunomodulating Therapy
Lenalidomide: Deletion 5q, or del(5q), is the most common cytogenetic abnormality in MDS, occurring in 10% to 15% of the MDS population. In general, del(5q) alone confers low-risk disease, but it is associated with significant anemia. Thalidomide was the first immunomodulatory agent studied for the treatment of MDS. Although it produced modest response rates, its use was limited owing to severe toxicity.1
Lenalidomide, a more potent derivative of thalidomide, was approved in 2005 for the treatment of patients with transfusion-dependent anemia due to lower-risk MDS associated with del(5q) with or without additional cytogenetic abnormalities. The mechanism of action of lenalidomide is not completely understood, but, like thalidomide, the drug is thought to exert immunomodulatory and antiangiogenic properties.7
The approval of lenalidomide for MDS was based on results of a phase II multicenter trial in which 148 patients with transfusion-dependent anemia and lower-risk MDS (IPSS Low or Int-1) associated with del(5q) with or without additional cytogenetic abnormalities received lenalidomide 10 mg orally daily (amended dose).11 Lenalidomide produced high response rates: 76% of patients achieved reduced transfusion needs and 67% became transfusion-independent. Responses were seen despite the number of cytogenetic abnormalities. The median time to response was 4.6 weeks; the median duration of response was not reached at 104 weeks’ follow-up. Of note, 45% of patients had a complete cytogenetic response, indicating that lenalidomide at least temporarily exerts a direct effect on the abnormal clone.11 To evaluate the efficacy and safety of lenalidomide in patients who were similar to those in the other trial but without del(5q), Raza and colleagues treated 214 patients with lenalidomide 10 mg daily.12 Compared with patients with del(5q), fewer hematologic responses were observed: 17% of patients experienced a >50% reduction in transfusion requirements and 26% became transfusion-independent.12
Lenalidomide has a favorable side-effect profile, with the exception of myelosuppression. Severe (grade 3/4) neutropenia and thrombocytopenia, the most common adverse events associated with lenalidomide, occurred in approximately half of patients with del(5q). Furthermore, myelosuppression resulted in dose interruption and/or reduction in 84% of patients.11 Myelosuppression related to lenalidomide typically arises within the first 8 weeks of treatment; therefore, it is recommended to check CBCs weekly during the first 8 weeks of therapy and at least monthly thereafter. Nonhematologic toxicities consisting of rash, pruritus, diarrhea, and fatigue were predominately grade 1/2.7,11 Lenalidomide is not associated with the severe somnolence, constipation, or neuropathy seen with thalidomide. Unlike its use in multiple myeloma, thrombosis occurrence has not been significant among MDS patients taking lenalidomide. Owing to the severe teratogenicity associated with thalidomide, lenalidomide is available only under a restricted program through the manufacturer.7 Under this program, called RevAssist, physicians, pharmacists, and patients must be registered in order to prescribe, dispense, or take lenalidomide, respectively. Strict patient counseling on the use of birth control, as well as routine monitoring, is required.
Lenalidomide has proven to be a beneficial treatment for transfusion-dependent anemia that is due to IPSS Low or Int-1 risk MDS associated with del(5q). Although it is not approved for non-del(5q) MDS, lenalidomide remains an option for patients with lower-risk MDS without del(5q) who remain transfusion-dependent after supportive-care measures and for patients who are not candidates for ESAs or more intensive therapy.
DNA Methyltransferase Inhibitors
Azacitidine and decitabine, both nucleoside analogues, were originally developed as cytarabine derivatives to be used at high doses for the treatment of leukemia. However, their use was limited owing to prolonged myelosuppression. After being reevaluated at lower doses, these agents were shown to work via epigenetic modulation as opposed to conventional cytotoxicity. In many cancers, including MDS, DNA hypermethylation is responsible for silencing the genes responsible for cell apoptosis and differentiation. At low doses, azacitidine and decitabine inhibit DNA methyltransferase, the enzyme responsible for DNA methylation, thereby restoring normal hematopoiesis.13 This new class of drugs is predominately used to treat patients with higher-risk MDS and lower-risk patients who are nonresponsive.
Azacitidine: The first hypomethylating agent approved by the FDA (in 2004), azacitidine is the only drug for MDS that is associated with increased OS. It is indicated for the treatment of all FAB subtypes of MDS. The pivotal trial that led to the drug’s approval was a phase III multicenter, randomized trial led by the Cancer and Leukemia Group B (CALGB).14 In this trial, 191 patients with all subtypes of MDS were randomized to receive azacitidine (75 mg/m2 subcutaneously [SC] daily x 7 days every 28 days) or best supportive care (BSC). Patients with progressive disease receiving BSC were allowed to cross over to the azacitidine arm. Compared with patients receiving BSC, those receiving azacitidine had significantly higher response rates by CALGB criteria (60% overall response rate [ORR], 7% complete response [CR], 16% partial response [PR], and 37% hematologic improvement [HI] vs. 5% HI for BCC; P <.0001). The median time to AML transformation or death was 21 months for azacitidine versus 13 months for BSC (P = .007). However, there was no significant difference in OS, likely owing to the study’s crossover design. The median time to response was 3 cycles, with a median duration of response of 15 months.14 When the data were reanalyzed using the WHO classification and International Working Group (IWG) response criteria, the ORR for azacitidine was 47% (10% CR, 1% PR, 36% HI) versus 17% HI for BSC.15-17 Most patients had a response by the 6th cycle of azacitidine.17
Subsequently, the efficacy of azacitidine was evaluated in a phase III trial that randomized 358 patients with higher-risk MDS to treatment with azacitidine at the approved dosing regimen or conventional care (intensive chemotherapy, low-dose cytarabine, or BSC).18 The primary outcome was OS. After a median follow-up of 21.2 months, the median survival was significantly longer with azacitidine compared with conventional care (24.5 vs. 15 months; hazard ratio [HR] 0.58; 95% CI 0.43-0.77; P = .0001). At 2 years, 51% of azacitidine-treated patients were alive versus 26% of conventional-care patients (P <.0001), and the median time to AML transformation was 17.8 months for azacitidine versus 11.5 months for conventional care (HR 0.5; 95% CI 0.35-0.7; P <.0001). By IWG criteria, an ORR of 77% (17% CR, 12% PR, 49% HI) was reported in the azacitidine arm versus 41% (8% CR, 4% PR, 29% HI) in the conventional-care arm.18 Notably, patients received a median of 9 cycles of azacitidine.18
Azacitidine is well tolerated overall, with myelosuppression being the most common toxicity.8,14,18 Myelosuppression usually occurs early in the course of treatment and can be complicated by the disease itself. Patients generally require several courses of therapy before a response is seen. For that reason, it is important to continue therapy for at least 4 cycles, in a timely fashion and despite cytopenias in the setting of persistent disease, before considering treatment failure. Neutropenia and thrombocytopenia tend to resolve with continued therapy and response to treatment. Growth factors can be utilized for neutropenia. The most common nonhematologic toxicities reported include injection-site reactions with SC administration, diarrhea, nausea, and vomiting.8 Patients should receive prophylaxis with a 5-hydroxytriptamine3–based antiemetic regimen. Although azacitidine has the convenience of outpatient administration, it entails weekend administration that often is limited in the community setting. Different schedules of azacitidine that eliminate weekend administration have been studied, but none has been directly compared with the FDA-approved regimen or has shown improved OS.
Decitabine: This agent was approved in 2006 for the treatment of all FAB subtypes of MDS with IPSS risk scores of Int-1, Int-2, and High. Decitabine has been evaluated at two different dosing regimens: the FDA-approved regimen of 15 mg/m2 IV over 3 hours every 8 hours for 3 consecutive days (135 mg/m2/cycle) every 6 weeks; and a lower dose of 20 mg/m2 IV over 1 hour daily for 5 days (100 mg/m2/cycle) every 4 weeks.
The phase III Registration Trial randomized 170 patients with all subtypes of MDS to treatment with BSC or decitabine 135 mg/m2/cycle every 6 weeks.19 The majority of decitabine patients had disease risk scores of IPSS Int-2 or High. By IWG criteria, the ORR was significantly higher with decitabine versus BSC (30% ORR, 9% CR, 8% PR, and 13% HI vs. 7% HI; P <.001), but there was no significant difference in the median time to AML or death (12.1 vs. 7.8 months, respectively) or OS.19 A median of 3 cycles of decitabine was administered.19 A similar phase III randomized study of patients with higher-risk MDS was conducted by the European Organization for Research and Treatment of Cancer (EORTC) and the German MDS Study Group.20 This study randomized 233 elderly patients to treatment with BSC or decitabine 135 mg/m2/cycle. Similar response rates to the Registration Trial were found, as well as an improvement in progression-free survival with decitabine; however, there was no difference in OS or time to AML. Almost half of patients in the decitabine arm received 2 cycles only.20
To maximize hypomethylation, a phase II study was conducted to evaluate the efficacy of decitabine at lower dose (100 mg/m2/cycle) schedules: 20 mg/m2 IV daily for 5 days; 20 mg/m2 (10 mg/m2 twice daily) SC daily for 5 days; or 10 mg/m2 IV daily for 10 days every 4 weeks regardless of counts (as long as there was persistent disease and no significant toxicity due to myelosuppression).21 An ORR of 73% by modified IWG criteria was reported.21 Patients who received 20 mg/m2 IV for 5 days had the best response rates, with 39% CR. This arm also was associated with the best degree of hypomethylation. A median of 9 cycles was given.21 Subsequently, another phase II study treated 99 MDS patients with decitabine 20 mg/m2 IV for 5 days every 4 weeks.22 Similar response rates were shown, with an ORR of 51% by modified IWG criteria.23 Additionally, the median survival was 19.4 months. A median of 5 cycles was given, and the majority of patients had a response by the 2nd cycle.22 This study confirmed the efficacy of lower-dose decitabine.
Similarly to azacitidine, decitabine is generally well tolerated. Myelosuppression—specifically, neutropenia and thrombocytopenia—is the most common toxicity.19-22 Growth factors may be used to prevent or shorten the duration of neutropenia in order to maintain dose intensity. The most common nonhematologic toxicities are hyperbilirubinemia, pyrexia, and pneumonia.9 As most patients require at least 3 or 4 cycles before response, therapy should be continued every 4 to 6 weeks (depending on the dosing schedule) and (as long as disease is present and there is no toxicity due to myelosuppression) for a minimum of 4 cycles before decitabine is stopped. Treatment with decitabine should continue as long as the patient shows benefit or there is no unacceptable toxicity.
CONCLUSION
The treatment of MDS has changed dramatically over the past decade. This is due in part to a better understanding of the disease and improved classification systems for MDS, as well as to the introduction of lenalidomide, azacitidine, and decitabine. Whereas lenalidomide has a clear role in the treatment of lower-risk MDS patients with del(5q) and transfusion-dependent anemia, azacitidine and decitabine are predominately used for higher-risk MDS patients. Azacitidine and decitabine appear to produce similar response rates; however, they have never been compared in a head-to-head randomized clinical trial. Recently, azacitidine was the first and only treatment for MDS to yield an improved OS. Despite advances in the treatment of MDS, complete remission rates and OS remain relatively low. Thus, the search for novel combinations of the approved agents, along with investigational agents, is underway.
REFERENCES
1. Faderl S, Kantarjian H. Myelodysplastic syndromes. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:2144-2154.
2. Sekeres MA, Schoonen WM, Kantarjian H, et al. Characteristics of US patients with myelodysplastic syndromes: results of six cross-sectional physician surveys. J Natl Cancer Inst. 2008;100:1542-1551.
3. Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007;109:1536-1542.
4. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079-2088.
5. Greenberg P, Cox C, LeBeau MM, et al. Erratum. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1998;91:1100.
6. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Myelodysplastic syndromes. V2.2010. www.nccn.org/professionals/physician_gls/pdf/mds.pdf. Accessed January 15, 2010.
7. Revlimid (lenalidomide) package insert. Summit, NJ: Celgene; January 2009. www.revlimid.com/pdf/REVLIMID_PI.pdf Accessed January 20, 2010.
8. Vidaza (azacitidine) package insert. Summit, NJ: Celgene; August 2008. www.vidaza.com/pdf/PI_FINAL.pdf. Accessed January 20, 2010.
9. Dacogen (decitabine) package insert. Woodcliff Lake, NJ: Eisai; March 2010. www.eisai.com/pdf_files/DacogenNewPI.pdf Accessed January 20, 2010.
10. Hellström-Lindberg E, Malcovati L. Supportive care, growth factors, and new therapies in myelodysplastic syndromes. Blood Rev. 2008;22:75-91.
11. List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456-1465.
12. Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1–risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111:86-93.
13. Garcia-Manero G. Modifying the epigenome as a therapeutic strategy in myelodysplasia. Hematology Am Soc Hematol Educ Program. 2007:405-411.
14. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the Cancer and Leukemia Group B. J Clin Oncol. 2002;20:2429-2440.
15. Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting—Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835-3849.
16. Cheson BD, Bennett JM, Kantarjian H, et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood. 2000;96:3671-3674.
17. Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895-3903.
18. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223-232.
19. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794-1803.
20. Wijermans P, Suciu S, Baila L, et al. Low dose decitabine versus best supportive care in elderly patients with intermediate or high risk MDS not eligible for intensive chemotherapy: final results of the randomized phase III study (06011) of the EORTC Leukemia and German MDS Study Groups. Blood. 2008;112:abstract 226.
21. Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of three schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52-57.
22. Steensma DP, Baer MR, Slack JL, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27:3842-3848.
23. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419-425.
To comment on this article, contact rdavidson@uspharmacist.com.



