US Pharm. 2010;35(9)(Oncology suppl):8-16.
ABSTRACT: Melanoma is the fifth most common cancer in the United States. In 2009, it was estimated that approximately 8,650 patients would die from melanoma. Numerous genetic and environmental risk factors have been studied, with a new focus on molecular mechanisms of disease progression. Current treatments include excisional surgery and adjuvant therapy for advanced melanoma and systemic chemotherapy and biochemotherapy for metastatic disease. Continued research on metastatic melanoma has provided new venues for potential therapies. Several treatment candidates have emerged over the last decade, including vaccines, angiogenesis inhibitors, and monoclonal antibodies.
Melanoma, the most serious form of skin cancer, has one of the fastest-growing incidence rates in the United States. In 1960, 1 in 1,500 people was expected to develop melanoma. In 2000, that ratio decreased to 1 in 70, and again to 1 in 50 in 2009. Currently, approximately 1 in 50 Caucasian, 1 in 200 Hispanic, and 1 in 1,000 African American individuals in the U.S. will develop melanoma in his or her lifetime.1
The number of people newly diagnosed with melanoma has increased over the last 10 years. In 1999, 44,200 patients were diagnosed, and 7,300 died from the disease. The American Cancer Society estimated that 68,720 new melanoma cases were diagnosed in 2009, with approximately 8,650 deaths attributed to the disease.2 The incidence is estimated to be rising by almost 6% per year, with the reported incremental cost ranging from approximately $4,650 for in situ tumors to about $160,000 for stage IV melanoma.3 In the U.S., the cost for treating melanoma in patients over 65 years of age was approximately $390 million.4
RISK FACTORS
A number of risk factors have been identified that play a role in the etiology of melanoma (TABLE 1).5 The combination of environmental factors and genetic factors such as gender and ethnicity creates a more complex picture of the development of melanoma. Family history is an important factor in the diagnosis of melanoma, as are personal characteristics such as blue eyes, fair or red hair, pale complexion, and a proclivity for sunburning and freckling. If a patient has at least one affected relative, his or her risk increases 2.2-fold; if two or more relatives have dysplastic nevi and/or melanoma, the risk is almost 100%.6 Patients exhibiting an increased number of benign and/or dysplastic melanocytic nevi are at higher risk for melanoma. Patients who are immunosuppressed are also at increased risk. Environmental risk factors include ultraviolet (UV) exposure and geographic location.

Several genes related to melanoma risk have been identified based on melanoma subclasses, anatomical site, extent of UV exposure, and mutational events of several genes.7 Genes such as CDKN2A and Ink4a/Arf and the Ras/Raf/MAPK pathway play a role in melanoma development.8 Similarly, BRAF and N-Ras mutations have been implicated in the development of the disease and may provide future targets for therapy.9 Current studies continue to explore the interplay between molecular genetics and the increased risk of melanoma in selected patients.
PATHOPHYSIOLOGY
The classical clinical presentation of melanoma varies by type, but a mole may be assessed according to the ABCDE mnemonic: Asymmetry; Border irregularity; Color variability or recent color change; Diameter (increasing, or >6 mm); and Evolving lesion, including surface changes (i.e., crusting, bleeding) or symptomatic presentation (i.e., itching, tenderness).9 A comprehensive skin examination by a dermatologist is critical for evaluating and monitoring the patient with multiple or atypical nevi, a history of excessive sun exposure, or cutaneous skin cancer or melanoma.
CLASSIFICATION AND STAGING
The American Joint Committee on Cancer (AJCC) staging system for melanoma was developed by examining the thickness of the primary tumor; ulceration of the lesion; and metastases to the lymph nodes, distant sites, and other organs. The TNM staging system is based on three characteristics that define extent of disease: tumor size (T), lymph node involvement (N), and metastasis (M). Stages I and II are localized to a specific region. Stage III, which encompasses the involvement of regional lymph nodes, is considered to be locoregional disease, and stage IV indicates distant sites of disease (e.g., brain, liver). TNM status and stage correlate to prognosis and survival.10,11 Tumor thickness and ulceration are the most powerful predictors of survival.12 Other predictors include localization, tumor markers, regression of the primary melanoma, and serum lactic dehydrogenase levels.12
PREVENTION
Public health awareness is one of the key factors in preventing melanoma, and identification of at-risk individuals can assist in early disease detection. High-risk patients should receive a yearly clinical examination, which has been shown to be cost-effective and yields improved survival. The American Academy of Dermatology recommends monthly skin self-examinations to identify moles or lesions that may be cancerous. The examination, performed using a mirror, should encompass all sides of the body and include the forearms, upper arms, palms, lower back, and feet (including interdigits). The American Cancer Society's recommendations for prevention include limiting direct sun exposure between the hours of 10 am and 4 pm, when UV rays are the most intense; wearing a broad hat to cover the face, as well as clothing that covers the arms, legs, and torso; and avoiding tanning beds.13 Applying sunscreen to exposed skin is also important; an SPF of 15 or higher, with protection against UVA and UVB rays, is recommended.
TREATMENT
Treatment options for the management of cutaneous melanoma are determined by the stage of disease at time of diagnosis. Surgical resection is based on the predicted risk of local recurrence and metastatic disease, as well as on the potential morbidity of the operation. If the lesion has not spread beyond the primary site, cure is possible, with an approximate 5-year survival rate of 90%.14 Most cutaneous lesions are thin (<1 mm). Clinical margins are based on the original thickness of the tumor; lymph node mapping and sentinel lymph node biopsy are suggested if the lesion is >1 mm.
Surgery remains the primary treatment modality for early-stage melanoma. Stages IB and II require larger excisions, with the potential for adjuvant therapy if the lesion is ³4 mm. Stage III requires lymph node dissection and adjuvant therapy. Follow-up biopsy with fine-needle aspiration of lymph nodes is recommended for progressive stage III disease. Use of adjuvant therapy in this setting depends upon the size of the mass, number of affected lymph nodes, and extension of the tumor. Other therapies include isolated limb perfusion (ILP) and radiation.
ILP
ILP is a treatment option for patients with involved locoregional metastasis. In patients with unresectable locally advanced melanoma, ILP allows for the direct administration of chemotherapy (CT) into the affected limb with minimal toxicity, as compared with systemic CT. The circulation of blood to and from the affected limb is temporarily stopped with a tourniquet, thereby preventing systemic circulation, while CT is administered through an extracorporeal pheresis system.15
A recent meta-analysis compared ILP with melphalan (with or without tumor necrosis factor [TNF] alfa) and evaluated clinical response, survival, and toxicity.16 Median complete response (CR) to ILP was 58.2 %, with an overall survival (OS) of 90.3%. An increase in overall response rate (ORR) was seen with melphalan + TNF-alfa. Reported rates for regional toxicity with ILP were 73.5%, 17.1%, and 2% for grades 2, 3, and 4, respectively. Women were at higher risk for toxicity.
Adjunctive Therapy
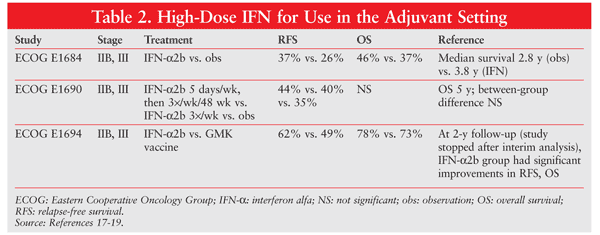
High-dose interferon (IFN) alfa is the only adjuvant therapy approved for high-risk melanoma patients, but it has never been prospectively tested in patients after resection of stage IV melanoma. (Note: stages IIB, IIC and III are considered high risk for adjuvant treatment.) Three Eastern Cooperative Oncology Group (ECOG) studies evaluated high-dose IFN-alfa in patients at high risk for recurrence (TABLE 2).17-19 Patients had stage IIB/C or III disease and received high-dose IFN-alfa after resection. OS was not affected by IFN-alfa therapy; however, the relapse-free interval was significantly prolonged. Whether the ECOG data are applicable in the setting of complete resection of distant metastases is currently unknown.20 High-dose IFN-alfa may be associated with significant toxic or adverse effects (e.g., liver toxicity), and some patients require dose reductions or therapy cessation owing to poor tolerance.
METASTATIC MELANOMA
Metastatic melanoma (MM) presents a true treatment challenge. In most studies, median survival is short, generally ranging from 6 to 9 months. CRs and durable responses are rare, and median survival has not greatly improved despite more than 30 years' experience with multimodal regimens.21
Single-Agent CT
Dacarbazine (DTIC) and Temozolomide (TMZ): In three large series performed in the early 1970s, response rates (RRs) for single-agent DTIC were 16%, 20%, and 28%, respectively; subsequent single-agent trials consistently produced an RR of less than 20% and a CR of 5%. Single doses of DTIC range from 800 mg/m2 to 1,000 mg/m2 every 21 days. DTIC has been the foundation for multidrug regimens despite its low activity as a single agent.22
TMZ is an oral alkylating agent, structurally related to DTIC, that is rapidly converted to the active metabolite MTIC. Unlike the case with DTIC, this conversion is nonenzymatic and spontaneous and occurs under physiologic conditions in all tissues to which it distributes.23 Activity of single-agent TMZ has been established in several phase I and II studies.
In a randomized trial of 305 patients with MM, median survival was greater--although not significantly (95% CI, 0.92-1.52)--with TMZ versus DTIC (7.7 vs. 6.4 months, respectively).24 TMZ was at least equivalent to DTIC in terms of time to progression (TTP), ORR, disease-free survival, and OS. TMZ was generally well tolerated, and the incidence of nausea and vomiting was similar between groups when prophylactic serotonin (5-HT3) antagonists were used. The trial was powered to detect a 50% increase in OS versus DTIC (superiority), but did not achieve its goal; thus, the FDA chose not to grant TMZ an indication for the treatment of melanoma.
One trial investigated the role of TMZ in the treatment of brain metastases.25 Among 155 patients, only 1 had CR, while 36% were deemed to have clinical benefit (CR + partial response [PR] + standard deviation). OS was a median of 3.2 months and was longer in patients who were CT-naïve versus those who were pretreated (3.5 vs. 2.2 months, respectively).
Other Single Agents: The platinums (carboplatin, cisplatin) and nitrosoureas (carmustine [BCNU], lomustine, semustine, fotemustine) are other classes that have shown single-agent activity.26 Cisplatin monotherapy induced a 15% RR and a duration of response (DR) of 3 months.25 A trial with carboplatin in 26 CT-naïve patients achieved an ORR of 19%.27 Trials with nitrosoureas have yielded similar results, with ORRs of 13% to 18%.25
Combination CT
In an attempt to improve RRs over single-agent therapy, several combination CT regimens have been evaluated. Cisplatin, vindesine, and semustine have been studied in combination with DTIC. None of the combinations fared better than single-agent DTIC, with RRs of 10% to 20%.28-30 Two of the most-studied combinations are the Dartmouth and CVD regimens.
Dartmouth Regimen: Also called CBDT, the Dartmouth regimen is a four-drug combination of cisplatin, BCNU, DTIC, and tamoxifen that was initially reported on in 1984. In 141 patients, the ORR was 46% with a median DR of 7 months, and the authors stated that the use of tamoxifen was essential.31 In a subsequent trial comparing CBDT with cisplatin, BCNU, and DTIC, CR was twice as high in the regimen without tamoxifen.32 In another randomized phase III trial, the Dartmouth regimen was compared with single-agent DTIC at 1,000 mg/m2 in 240 patients.33 Median survival was 1.4 months longer in the Dartmouth arm, and the ORR for Dartmouth compared with DTIC was 18.5% versus 10.2%, respectively. In the end, there was no improvement in OS with the Dartmouth regimen, with twice as much myelosuppression.
CVD Regimen: A phase II trial that added vinblastine and cisplatin to DTIC (CVD) was conducted to determine the regimen's antitumor effects in advanced melanoma patients who were not surgical candidates and were CT-naïve.34 Of 50 evaluable patients, 4% achieved CR, with an ORR of 40% (95% CI, 27%-55%). Notably, 53% of patients with liver metastases showed a response, which is uncommon with single-agent DTIC. TTP in patients considered to be responders was 9 months; however, 12 of the 22 patients who had any type of response (CR + PR + minimal response) subsequently developed brain metastases. A later study that compared CVD with DTIC in nearly 150 patients found no difference in DR or survival.35
Biotherapy (BIO)
The natural course of melanoma suggests a role for immunomodulation in treatment.36 The best-studied entities are the cytokines IFN-alfa and interleukin-2 (IL-2), which are often used in combination. Multiple trials exist, with CR of approximately 10% and an ORR of 40% to 60%.37-39
High-Dose Bolus IL-2: IL-2 has no direct effect on tumor cells; rather, its antitumor activity lies in modulating immunologic reactivity.40 IL-2 is FDA-approved for high-dose bolus administration in MM, but it also is used off-label in conjunction with IFN-alfa and CT. High-dose IL-2 administration induces durable CR in a small subset of patients, but it does so at the expense of hospital admission and severe adverse effects. A consecutive series assessed 283 patients with MM and renal cell carcinoma (RCC) who were treated with high-dose IL-2.40 CR was achieved in 7% of the 134 melanoma patients, with an ORR of 23%. Maximum duration of CR extended beyond 91 months. A meta-analysis examining 270 MM patients noted similar results, including an ORR of 16%.41 Six percent of patients achieved CR and progression-free survival (PFS) of at least 3 months. Toxicity was severe, but rapidly reversible; 6 deaths were attributable to treatment-related sepsis, however.
Biochemotherapy (BCT): In one study, IFN-alfa and IL-2 (BIO) were added to CVD and the regimens were tested in both alternating and sequential fashion.37 Overall, one of the sequential BCT arms (CVD/BIO) versus CVD alone demonstrated increased CR, prolonged TTP (nonsignificant [NS]), and an increase in median survival (P = .04.) Versus the BIO/CVD regimen, CVD/BIO exhibited a higher percentage of CRs and a significantly longer TTP (P = .02). CRs in both sequential groups were thought to be superior in quality, as they were observed in areas of bulky disease and were durable (³3-5 years). The sequential regimens exhibited more severe toxicities (i.e., hematologic), with nearly two-thirds of patients experiencing grade 4 thrombocytopenia. Febrile neutropenia developed in 71% of patients, with 50% of these yielding a positive blood culture. Other trials attempted alterations in the BCT regimen that were aimed at less toxicity and outpatient administration.42
Several phase III trials and three meta-analyses have examined various combinations of CT, BIO, and BCT. In the first meta-analysis, 631 patients were treated with four regimens (IL-2 alone, IL-2/CT, IL-2/IFN-alfa, or IL-2/IFN-alfa/CT), with ORRs of 15%, 21%, 23%, and 45%, respectively.43 The longest median survival was 11.4 months in the IL-2/IFN-alfa/CT group, which translated to a 12% survival rate at 5 years. Despite the RRs, median survival between groups was not significantly different. The second meta-analysis, involving 7,711 patients, revealed that treatment with IL-2/IFN-alfa/CT demonstrated a significant improvement in ORR versus CT or BIO alone.44 The most recent meta-analysis compared trials using CT with or without IFN-alfa and CT with or without IL-2/IFN-alfa.45 Patients who received BIO (IFN-alfa or IL-2/IFN-alfa) had significant benefit in terms of CR, PR, and ORR, but there was no difference in DR and no survival benefit between arms.
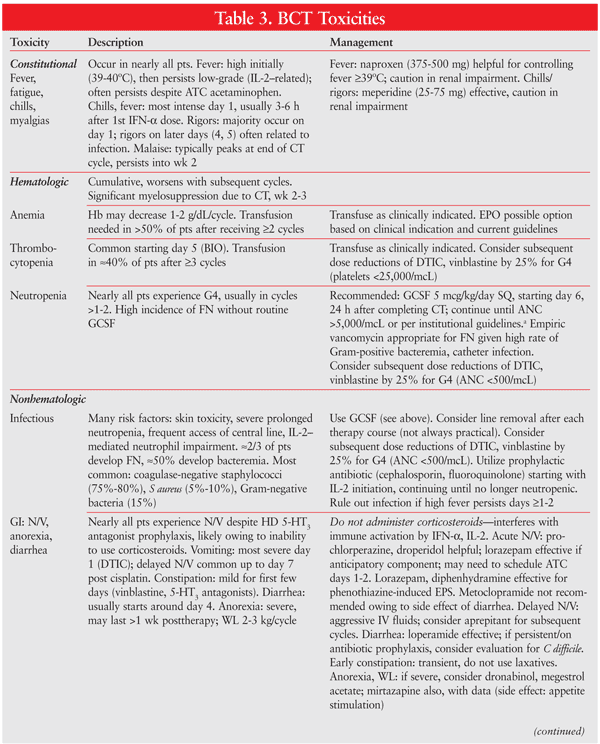
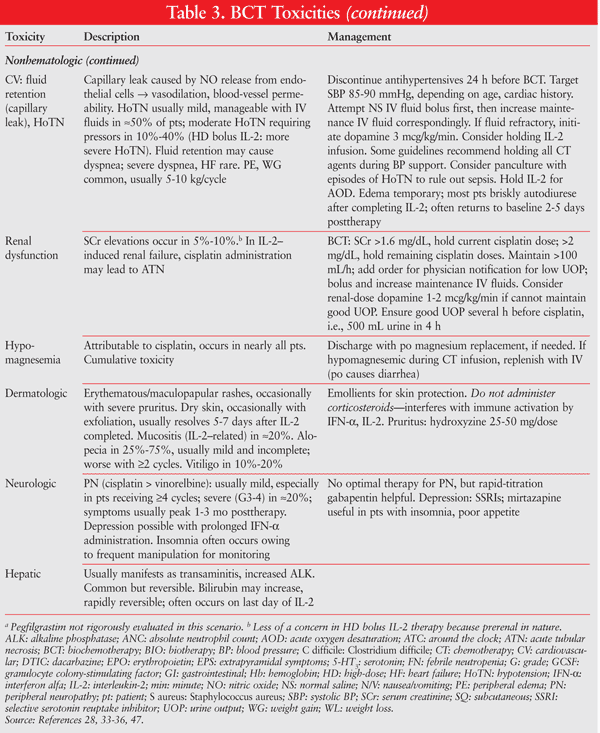
An ECOG-coordinated trial of 395 patients studied CVD versus CVD/IL-2/IFN-alfa.46 Median survival was 5.7 months longer in the BCT arm (19.5 months vs. 13.8 months; NS) and OS differed by only 0.3 months between arms. However certain subsets of patients (performance status 0, males) receiving BCT experienced a significant benefit in PFS, while other subsets (AJCC stage M1c, no prior IFN-alfa) had significantly lower PFS with BCT. See TABLE 3 for the management of BCT toxicities.6,47
INVESTIGATIONAL AGENTS
Targeted therapies have been a focal point in melanoma research. Studies of several antiangiogenesis agents and tyrosine kinase inhibitors have shown promise in preclinical and early-phase clinical trials. Sorafenib is a multikinase inhibitor, targeting CRAF, vascular endothelial growth factor receptor 2, platelet-derived growth factor beta, p39, flt-3, and c-kit.48 While sorafenib is currently approved for use in RCC and hepatocellular carcinoma, several phase I and II studies in melanoma have been performed, most of which have demonstrated stable disease.
Immunologic therapy continues to be explored as a possible treatment for melanoma. Research efforts have focused on vaccines derived from tumor cells, DNA, peptides, and dendritic cells, as well as on different emulsions for adjuvants to potentiate immune responses. Numerous early-phase clinical trials have shown that vaccines are able to stimulate the immune system in patients with metastatic disease.49 However, these trials involved a limited number of patients; larger trials are needed to make further conclusions about efficacy in metastatic and adjuvant settings.
Newer treatment options are in later stages of development. Research on immune regulation via cytotoxic T-lymphocyte antigen (CTLA) 4 protein on activated T cells shows promise. Melanoma has been shown to be an immunogenic tumor that may be affected by enhancing the immune system. The premise of using monoclonal antibodies (mAbs) against CTLA protein is to inhibit CTLA-4 cell expression, thereby allowing for upregulation of the immune system to target melanoma cells.50,51 Two different mAbs, ipilimumab and tremelimumab, have been progressing through late-stage clinical trials.
Ipilimumab is an immunoglobulin G (IgG) 1 mAb directed against the CTLA-4 protein. In recent clinical studies, ipilimumab has shown efficacy in combination with other CT agents and vaccines, but also as monotherapy. Responses to ipilimumab often occur several weeks after treatment. Side effects include rash, colitis, hypophysitis, uveitis, vitiligo, and infusion-related reactions.52,53
Another CTLA antibody, tremelimumab (CP-675,CP-206)--an IgG2 fully human mAb--is being investigated. Initial studies showed a durable response in patients with metastatic disease. Autoimmune effects include vitiligo, dermatitis, thyroiditis, hypophysitis, and colitis.54
SUMMARY
Melanoma is a complicated disease that predominately affects adults later in life. It remains one of the few malignancies with an increasing rate of morbidity and mortality. Surgery remains one of the primary treatment modalities, in conjunction with adjuvant therapies for patients with late-stage disease. MM treatment varies based on institutional protocols and patient characteristics. Therapy may include single-agent or combination CT, BIO, and BCT, all of which are effective in certain patient populations and have extensive toxicity profiles. Future trials of melanoma therapy are focused on immunotherapy and vaccine development. Continued research in this field will help determine the optimal treatment for melanoma and potentially enable the introduction of new classes of anticancer medications.
REFERENCES
1. Jemal A, Murray T, Samuels A, et al. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5-26.
2. American Cancer Society. Cancer facts & figures 2009.
www.cancer.org/Research/
3. Alexandrescu DT. Melanoma costs: a dynamic model comparing estimated overall costs of various clinical stages. Dermatol Online J. 2009;15:1.
4. Seidler AM, Pennie ML, Veledar E, et al. Economic burden of melanoma in the elderly population: population-based analysis of the Surveillance, Epidemiology, and End Results (SEER)--Medicare data. Arch Dermatol. 2010;146:249-256.
5. Ferrone CR, Ben Porat L, Panageas KS, et al. Clinicopathological features of and risk factors for multiple primary melanomas. JAMA. 2005;294:1647-1654.
6. Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma: a global perspective. Arch Dermatol. 2001;137:1493-1496.
7. Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135-2147.
8. Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22:3053-3062.
9. Rigel DS, Friedman RJ, Kopf AW, Polsky D. ABCDE--an evolving concept in the early detection of melanoma. Arch Dermatol. 2005;141:1032-1034.
10. Markovic SN, Erickson LA, Rao RD, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc. 2007;82:364-380.
11. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
12. Markovic SN, Erickson LA, Rao RD, et al. Malignant melanoma in the 21st century, part 2: staging, prognosis, and treatment. Mayo Clin Proc. 2007;82:490-513.
13. American Cancer Society. Cancer Prevention & Early Detection Facts & Figures 2006. Atlanta, GA: American Cancer Society; 2006.
14. Massi D, Franchi A, Borgognoni L, et al. Thin cutaneous malignant melanomas (£1.5 mm): identification of risk factors indicative of progression. Cancer. 1999;85:1067-1076.
15. Thompson JF, Kam PC. Current status of isolated limb infusion with mild hyperthermia for melanoma. Int J Hyperthermia. 2008;24:219-225.
16. Moreno-Ramirez D, de la Cruz-Merino L, Ferrandiz L, et al. Isolated limb perfusion for malignant melanoma: systematic review on effectiveness and safety. Oncologist. 2010;15:416-427.
17. Kirkwood JM, Strawderman MH, Ernstoff MS, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7-17.
18. Kirkwood JM, Ibrahim JG, Sondak VK, et al: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000;18:2444-2458.
19. Kirkwood J, Ibrahim JG, Sosman JA, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of Intergroup Trial E1694/S9512/C509801. J Clin Oncol. 2001;19:2370-2380.
20. Eggermont AMM, Testori A, Marsden J, et al. Utility of adjuvant systemic therapy in melanoma. Ann Oncol. 2009;20:vi30-vi34.
21. Gogas HJ, Kirkwood JM, Sondak VK. Chemotherapy for metastatic melanoma: time for a change? Cancer. 2007;109:455-464.
22. Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40:1825-1836.
23. Agarwala SS, Kirkwood JM, Gore M, et al. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: a phase II study. J Clin Oncol. 2004;22:2101-2107.24. Temozolomide. Hudson, OH: Lexi-Comp, Inc. Version 1.6.3.
25. Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18:158-166.
26. Costanza M, Nathanson L, Schoenfeld D, et al. Results with methyl-CCNU and DTIC in metastatic melanoma. Cancer. 1977;40:1010-1015.
27. Evans LM, Casper ES, Rosenbluth R. Phase II trial of carboplatin in advanced malignant melanoma. Cancer Treat Rep. 1987;71:171-172.
28. Atkins MB. The treatment of metastatic melanoma with chemotherapy and biologics. Curr Opin Oncol. 1997;9:205-213.
29. Fletcher WS, Green S, Fletcher JR, et al. Evaluation of cis-platinum and DTIC combination chemotherapy in disseminated melanoma. A Southwest Oncology Group Study. Am J Clin Oncol. 1988;11:589-593.
30. Vorobiof DA, Sarli R, Falkson G. Combination chemotherapy with dacarbazine and vindesine in the treatment of metastatic malignant melanoma. Cancer Treat Rep. 1986;70:927-928.
31. Del Prete SA, Maurer LH, O'Donnell J, et al. Combination chemotherapy with cisplatin, carmustine, dacarbazine, and tamoxifen in metastatic melanoma. Cancer Treat Rep. 1984;68:1403-1405.
32. Lattanzi SC, Tosteson T, Chertoff J, et al. Dacarbazine, cisplatin and carmustine, with or without tamoxifen, for metastatic melanoma: 5-year follow-up. Melanoma Res. 1995;5:365-369.
33. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17:2745-2751.
34. Legha SS, Ring S, Papadopoulos N, et al. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer. 1989;64:2024-2029.
35. Buzaid AC, Legha SS, Winn R, et al. Cisplatin (C), vinblastine (V), and dacarbazine (D) (CVD) versus dacarbazine alone in metastatic melanoma: preliminary results of a Phase II Cancer Community Oncology Program (CCOP) trial [abstract]. Proc Am Soc Clin Oncol. 1993;12:389a.
36. Bajetta E, Del Vecchio M, Bernard-Marty C, et al. Metastatic melanoma: chemotherapy. Semin Oncol. 2002;29:427-445.
37. Legha SS, Ring S, Bedikian A, et al. Treatment of metastatic melanoma with combined chemotherapy containing cisplatin, vinblastine and dacarbazine (CVD) and biotherapy using interleukin-2 and interferon-alpha. Ann Oncol. 1996;7:827-835.
38. Legha SS, Ring S, Eton O, et al. Development of a biochemotherapy regimen with concurrent administration of cisplatin, vinblastine, dacarbazine, interferon alfa, and interleukin-2 for patients with metastatic melanoma. J Clin Oncol. 1998;16:1752-1759.
39. Atkins MB, O'Boyle KR, Sosman JA, et al. Multiinstitutional phase II trial of intensive combination chemoimmunotherapy for metastatic melanoma. J Clin Oncol. 1994;12:1553-1560.
40. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell carcinoma using high-dose bolus interleukin-2. JAMA. 1994;271:907-913.
41. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-2116.
42. O'Day SJ, Gammon G, Boasberg PD, et al. Advantages of concurrent biochemotherapy modified by decrescendo interleukin-2, granulocyte colony-stimulating factor, and tamoxifen for patients with metastatic melanoma. J Clin Oncol. 1999;17:2752-2761.
43. Keilholz U, Conradt C, Legha SS, et al. Results of interleukin-2-based treatment in advanced melanoma: a case record-based analysis of 631 patients. J Clin Oncol. 1998;16:2921-2929.
44. Allen IE, Kupelnick B, Kumashiro M. Efficacy of interleukin-2 in the treatment of metastatic melanoma. Systematic review and meta-analysis. Cancer Ther. 1998;1:168-173.
45. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. J Clin Oncol.
46. Atkins MB, Hsu J, Lee S, et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2008;26:5748-5754.
47. Buzaid AC, Atkins M. Practical guidelines for the management of biochemotherapy-related toxicity in melanoma. Clin Cancer Res. 2001;7:2611-2619.
48. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-7109.
49. Kirkwood JM, Moschos S, Wang W. Strategies for the development of more effective adjuvant therapy of melanoma: current and future explorations of antibodies, cytokines, vaccines, and combinations. Clin Cancer Res. 2006;12:S2331-S2336.
50. Terando AM, Faries MB, Morton DL. Vaccine therapy for melanoma: current status and future directions. Vaccine. 2007;25(suppl 2):B4-B16.
51. Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611-618.
52. Weber J. Overcoming immunological tolerance to melanoma: Targeting CTLA-4 with ipilimumab (MDX-010). Oncologist. 2008;13(suppl 4):16-25.
53. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24:2283-2289.
54. Ribas A, Camacho LH, Lopez-Berestein G, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23:8968-8977. 2007;25:5426-5434.
To comment on this article, contact rdavidson@uspharmacist.com.


