US Pharm. 2011;36(11)(Oncology suppl):5-12.
ABSTRACT: Pharmacogenomics (PGx) plays a significant role in the pharmacotherapy of cancer, as narrow therapeutic indices, low overall response rates, rapid and severe systemic toxicity, and unpredictable efficacy are all hallmarks of cancer therapies. The implementation of pharmacogenomics in cancer treatment offers the potential for clinicians to better predict the differences in drug response, resistance, efficacy, and toxicity among chemotherapy and targeted-therapy patients, and to optimize the treatment regimens based on these differences. This review highlights the current and future potential therapeutic applications of selective PGx biomarkers in oncology.
According to the National Council on Patient Information and Education, adverse drug reactions (ADRs) to prescribed drugs are one of the leading causes of death in the United States.1 It is estimated that the overall incidence of serious ADRs occurred in 6.7% of hospitalized patients from 1966 to 1996, with over 2.2 million serious and 106,000 fatal ADRs in 1994 alone.1 Interracial, interethnic, and interindividual polymorphisms in genes that encode for drug-metabolizing enzymes, drug transporters, drug targets, and many other factors that affect the pharmacokinetics and/or pharmacodynamics of the pharmacotherapy have been linked to ADRs.2
The ultimate goal of pharmacogenomics (PGx) research is to reduce ADRs and improve therapeutic outcomes by utilizing genomic technologies to identify genetic polymorphisms in patients that make them more susceptible to developing certain diseases or impair the pharmacologic function of, and therapeutic response to, specific drugs. With this information, clinicians can correlate these specific inherited genetic differences with interindividual differential drug disposition and its effects to customize drug therapy regimens.3 In 2004, for example, the FDA approved the first pharmacogenetic test (AmpliChip CYP450 Test) to identify a patient’s CYP2D6 and CYP2C19 genotype by analyzing DNA extracted from a whole blood sample.4
Nowhere is PGx research needed more than in cancer treatment. Many chemotherapeutic agents have a narrow therapeutic index (NTI), low overall response rate, rapid and severe systemic toxicity, and unpredictable efficacy. The administration of the same standard dose anticancer drug to a population of patients results in a range of toxicity from unaffected to death. Accumulating evidence indicates that PGx therapy has already been successfully applied to predict cancer susceptibility, drug response, and toxicity associated with traditional chemotherapy treatments in specific patient populations.5 In addition, PGx data have also shed new light on the efficacy and toxicity of molecular targeted anticancer agents, including monoclonal antibody therapeutics.6
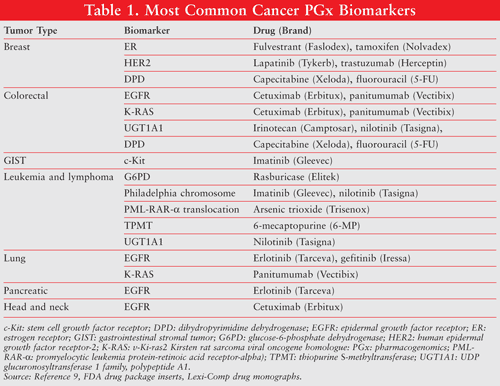
Over the past decade, PGx data have greatly affected all aspects of oncology research and practice. In 2005, the FDA issued guidelines for the submission of PGx research during the drug development process.7 Consequently, PGx information has been an important component of drug package inserts for many chemotherapeutic agents, including tamoxifen, 5-fluorouracil, irinotecan, trastuzumab, and cetuximab. In oncology, emerging clinical evidence from PGx studies has enabled health care professionals to use PGx indicators to predict tumor cell susceptibility to certain therapeutic agents and the patient’s prognosis after treatment, and to increase the drug response and decrease the dose-limiting toxicities associated with cancer treatment.8 TABLE 1 lists the PGx biomarkers associated with cancer treatments cited in FDA-approved drug labels.9 Common biomarkers include gene variants, functional deficiencies, expression changes, and chromosomal abnormalities. Most PGx biomarkers can be categorized as either selective biomarkers for clinical response, biomarkers for preventing toxicities, or biomarkers for drug resistance.10 This review will focus on the biomarkers for selective therapy.
PGx Biomarkers for Selection of Therapy
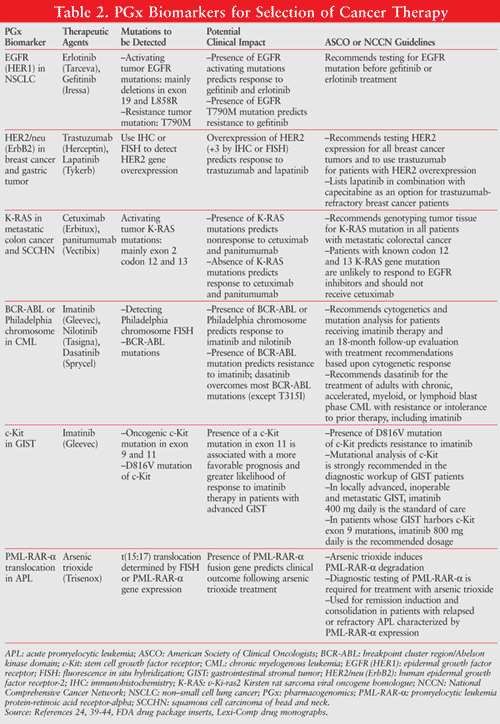
Biomarkers for selection of therapy are usually associated with targeted therapeutic agents directed at tumor cells with particular protein characteristics that significantly differ from their normal cell counterparts. By identifying specific PGx biomarkers present in tumors, physicians can select and tailor a patient’s treatment based on his or her genetic profile. Thus, targeted therapy guided by PGx biomarkers has the potential to be more selective for cancer cells than for normal cells, which can significantly improve the prognosis of cancer patients and potentially decrease the toxic effects of anticancer drugs on normal cells. Examples of these indicators include epidermal growth factor receptor (EGFR), K-RAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue), human epidermal growth factor receptor-2 (HER2), and stem cell growth factor receptor (c-Kit).5,11,12 TABLE 2 summarizes the key PGx biomarkers for selection of therapy cited by the FDA cancer drug labels for therapeutic indications with clinical evidence from PGx studies. These common PGx biomarkers play an important role in cancer treatment by identifying responders from nonresponders to medications, avoiding ADRs, and optimizing drug dose.
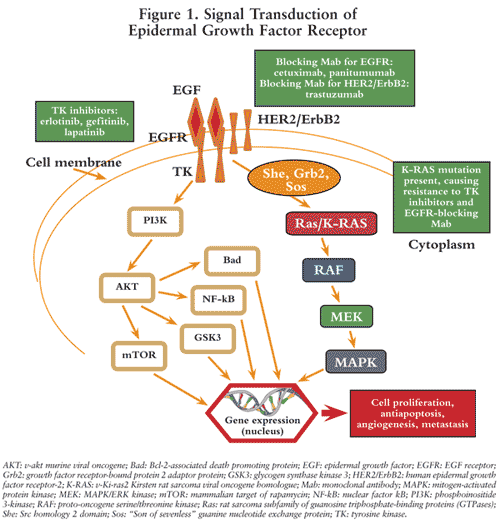
Epidermal Growth Factor Receptor (EGFR, HER1, c-ErbB1): This is a transmembrane glycoprotein that is a member of a subfamily of type I receptor tyrosine kinases including EGFR, HER2, HER3, and HER4. EGFR contains an extracellular ligand-binding domain, a cytoplasmic tyrosine kinase domain, and a single transmembrane domain.12 The extracellular domain is engaged in ligand binding and receptor homo- or hetero-dimerization. The dimerization triggers autophosphorylation of the c-terminus region of the tyrosine kinase domain and initiates the cascade of downstream signal transduction, including recruitment of the adapter proteins Grb2 and Sos, which bind to the receptor and activate Ras (rat sarcoma). Ras then activates the extracellular regulated kinase (ERK) and c-Jun NH2-terminal kinase (JNK) signal transduction pathways, which subsequently activate transcription factors leading to cell proliferation. The EGFR is constitutively expressed in many normal epithelial tissues. Overexpression of EGFR is also detected in many human cancers, including those of the head, neck, colon, and rectum. Therefore, EGFR gene overexpression is a strong indicator for selecting and predicting response to cetuximab, panitumumab, erlotinib, and gefitinib (FIGURE 1).13
Cetuximab is a recombinant, chimeric human/mouse monoclonal antibody that binds specifically to the extracellular domain of the human receptor (i.e., EGFR) and competitively inhibits the binding of epidermal growth factor (EGF) and other ligands, such as transforming growth factor–alpha.14 Panitumumab is a recombinant, human immunoglobulin (Ig)G2 kappa monoclonal antibody that also binds specifically to the human EGFR.15 Binding of cetuximab and panitumumab to the EGFR on tumor cells has been shown to block receptor phosphorylation and activation of receptor-associated kinases, resulting in inhibition of cell growth, induction of apoptosis, and decreased matrix metalloproteinase and vascular endothelial growth factor production.14,15 In contrast, erlotinib and gefitinib directly inhibit the intracellular phosphorylation of the tyrosine kinase associated with the EGFR (FIGURE 1).16,17 Although they share a similar mechanism of action, each of the above EGFR inhibitors has different drug label instructions based on the evidence from clinical trials and PGx data.12,16,17
Somatic mutations are alterations in DNA sequence that can occur in the gene-coding sequences (exons) that take place after conception, which may arise in any cell in the body. The sequence alterations can sometimes cause cancer or other diseases. A variety of somatic mutations in exons 18, 19, and 21 of EGFR gene corresponding to the tyrosine kinase domain of EGFR have been defined, especially deletion in exon 19 and the point mutation in exon 18 (G719A/C), which results in an amino acid substitution at position 719 in EGFR from a glycine (G) to an alanine (A) or to a cysteine (C). Another important point of mutation occurs in exon 21, which results in an amino acid substitution (L858R) at position 858 in EGFR from a leucine (L) to an arginine (R), or an amino acid substitution (L861Q) at position 861 from a leucine (L) to a glutamine (Q).
Somatic mutations in exons 18, 19, and 21 of the EGFR gene constitutively activate the EGFR tyrosine kinase domain, which triggers the cascade of signal transduction of EGFR without the ligand binding. These activating mutations are observed in 15% of Caucasians and 60% of Asians.18 Whereas the T790M mutation (amino acid substitution at position 790 in EGFR, from a threonine [T] to a methionine [M]) is present in a small subpopulation of tumor cells shown to be resistant to gefitinib or erlotinib, the presence of an EGFR-activating mutation in advanced stages of non–small cell lung cancer (NSCLC) treated with gefitinib or erlotinib increases the median survival from 10 to 27 months.19 In the absence of such EGFR-activating mutations, gefitinib therapy is not superior to conventional chemotherapy. The presence of the T790M resistance mutation at presentation, together with an EGFR-activating mutation, predicts a shorter time to progression of the disease, whereas the absence of EGFR-activating mutations is clearly associated with a nonresponse to gefitinib. In a phase III trial of erlotinib involving patients with progression after standard chemotherapy for NSCLC, Tsao et al demonstrated that patients without EGFR-activating mutations seem to have a slightly better outcome with erlotinib compared with a placebo.20
HER2/ErbB2: This is a membrane glycoprotein belonging to the EGF receptor family. Despite being unable to directly bind growth factors, HER2 forms a hetero-dimer with other ligand-bound EGF receptor family members, helping stabilize ligand binding and enhance kinase-mediated activation of downstream molecules. HER2 can be used in determining the prognosis of numerous carcinomas, including breast, prostate, ovarian, and lung cancers. HER2 is overexpressed in approximately one-fourth of breast cancer patients. Overexpression of the HER2 oncogene is correlated with a poor prognosis, increased tumor growth and metastasis, and resistance to chemotherapeutic agents.21
Trastuzumab is a recombinant DNA-derived humanized monoclonal antibody that selectively binds with high affinity to the extracellular domain of HER2. Assessment of HER2 status by immunohistochemistry (IHC) or fluorescence in situ hybridization (FISH) is the standard for the evaluation of newly diagnosed carcinomas of the breast. HER2 overexpression is an indicator necessary for selecting patients for trastuzumab therapy.22
Lapatinib is an oral dual tyrosine kinase inhibitor that targets EGFR and HER2. Lapatinib can inhibit HER2 activation via ligand-induced heterodimerization or truncated HER2 receptors. In a randomized phase III trial to study the efficacy and safety of lapatinib in patients with HER2-positive metastatic breast cancer who had experienced progression on prior trastuzumab therapy, Blackwell et al demonstrated that trastuzumab-refractory breast cancer patients responded to lapatinib, and lapatinib in combination with trastuzumab significantly improved progression-free survival compared to lapatinib alone.23 The National Comprehensive Cancer Network (NCCN) lists lapatinib in combination with capecitabine as an option for trastuzumab-refractory breast cancer patients.24
K-RAS: This is a Kirsten Ras oncogene homologue from the mammalian Ras gene family. This oncogene encodes a protein that is a member of the small GTPase (guanosine triphosphate-binding protein) superfamily, which plays an important role in the signal transduction of EGFR. The ligand binding to EGFR activates signal transduction and results in K-RAS wild-type (WT) activation. The oncogenic mutations of the K-RAS gene lead to the accumulation of Ras protein in the active GTP-bound state, which activate the downstream signal transduction pathway of EGFR without ligand binding.1 In addition to the mutations of the EGFR tyrosine kinase domain, mutated forms of K-RAS have been shown to be present in many human tumors, including colon and lung cancers. Approximately 97% of K-RAS mutations seen in NSCLC patients involve codons 12 and 13.25 K-RAS–activating mutations were shown to be a negative predictor for the response to cancer therapy with certain anti-EGFR monoclonal antibodies (panitumumab, cetuximab) and tyrosine kinase inhibitors (erlotinib, gefitinib).25
Evidence links K-RAS mutations to poor survival in colorectal cancer. In a phase III metastatic colorectal cancer (mCRC) trial, Amado et al demonstrated that the effect of panitumumab on progression-free survival (PFS) differed by K-RAS status. K-RAS mutation was associated with shorter PFS compared to WT (7.4 vs. 12.3 weeks) and to the control group with best supportive care. For the subgroup of patients with K-RAS mutation, no benefit was observed with the treatment of panitumumab compared to best supportive care.26 Van and his research group’s phase III trial data also indicated no benefit of adding cetuximab to irinotecan, fluorouracil, and leucovorin (FOLFIRI) as first-line treatment for mCRC in K-RAS mutation patients (median PFS = 7.6 vs. 9.9 months in mutant [MT] and WT patients, respectively; median overall survival [OS] = 17.5 vs. 24.9 months in MT and WT patients, respectively).27 Thus, anti-EGFR therapies are indicated only in patients determined to have K-RAS WT tumors.
In July 2009, the FDA added K-RAS testing information to the panitumumab and cetuximab labels stating that subset analyses of metastatic or advanced colorectal cancer trials have not shown a treatment benefit for either drug in patients whose tumors had K-RAS mutations in codon 12 or 13. Use of cetuximab or panitumumab is not recommended for the treatment of colorectal cancer with these mutations.28,29
Philadelphia Chromosome (Philadelphia Translocation): This is a specific chromosomal abnormality resulting from reciprocal translocation of genetic material between chromosomes 9 and 22, causing the formation of the oncogenic BCR-ABL (breakpoint cluster region/Abelson kinase domain) fusion gene. This gene encodes for BCR-ABL fusion protein that is an abnormal, constitutively active tyrosine kinase important for cell proliferation and apoptosis. Philadelphia chromosome is usually associated with chronic myelogenous leukemia (CML), with 95% of CML patients having this abnormality.30
Imatinib has revolutionized the therapy of newly diagnosed patients with CML by binding to the inactive configuration domain of BCR-ABL kinase and competitively inhibiting the adenosine triphosphate (ATP) binding site. Despite the success of imatinib therapy for most CML patients, some individuals may not achieve the desired response or may eventually lose an adequate response to imatinib due to mutations at the tyrosine kinase domain.31 Like imatinib, nilotinib also binds to the inactive conformation domain of BCR-ABL kinase. Therefore, nilotinib is not the best alternative treatment for most imatinib-resistant mutations. However, the novel small molecular tyrosine kinase inhibitor dasatinib binds to the kinase domain in the open conformation, which is different from the mechanism of action of imatinib and nilotinib. Thus, dasatinib can and should be reserved for CML patients who are resistant to imatinib and nilotinib. However, CML patients with the T315I mutation (threonine at position 315 mutated to isoleucine) are resistant to imatinib, nilotinib, and dasatinib.32-34
Other than for Philadelphia chromosome–positive (Ph+) CML, the FDA also approved imatinib for the c-Kit positive (CD117) gastrointestinal stromal tumors (GISTs) and aggressive systemic mastocytosis (ASM) without the D816V (aspartic acid at 816 mutated to valine) c-Kit mutation (or c-Kit mutation status unknown) based on the PGx studies (TABLE 2). Imatinib is a potent inhibitor for tyrosine kinase derived from both BCR-ABL and c-Kit.35,36
Like most CML patients who have Philadelphia translocation, about 98% of patients with acute promyelocytic leukemia (APL) possess the PML-RAR-a (promyelocytic leukemia protein-retinoic acid receptor-alpha) translocation, which creates a fusion protein that arrests promyelocytic differentiation. Normally, PML-RAR-a functions as a ligand-activated transcription factor associated with corepressors or coactivators, which repress or activate genes that control myeloid differentiation. PML-RAR-a fusion protein has increased binding ability to the transcriptional corepressors N-CoR (nuclear receptor corepressor) and SMRT (silencing mediator of retinoid and thyroid receptors), resulting in the abnormal constitutive silencing of RAR target genes, which arrest promyelocytic differentiation.37 Arsenic trioxide induces PML-RAR-a degradation and was approved by the FDA for remission induction and consolidation in patients with relapsed or refractory APL characterized by PML-RAR-a expression. Diagnostic testing for PML-RAR-a is required for treatment with arsenic trioxide.38
Conclusion and Prospects
The goal of pharmacogenomics is to elucidate the genetic basis for interindividual differences in drug response and to use this genetic information to predict the safety, toxicity, and efficacy of drugs in individuals or groups of patients. Although the therapeutic application of PGx research in cancer treatment during the past decade has been initially slow, the accumulating evidence of bench work and bedside patient care has been growing exponentially. By identifying specific PGx biomarkers present in tumors, physicians can select and tailor a patient’s treatment based on his or her genetic profile.
Furthermore, it has become clear that the clinical benefit associated with these therapeutic agents targeted at the specific biomarkers is typically limited to a subset of responsive patients with or without a specific genomic mutation of the biomarkers. Thus, targeted therapy guided by PGx biomarkers has the potential to significantly improve the prognosis of selective cancer patients and avoid the costly unresponsive treatment for the nonselective patients to save valuable time for other proper treatments. There is an unprecedented urgency for more clinicians to become trained on how to interpret data from PGx testing and to prepare for the upcoming future of health care—personalized medicine.
REFERENCES
1. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200-1205.
2. Shah RR. Pharmacogenetics in drug regulation: promise, potential and pitfalls. Philos Trans R Soc Lond B Biol Sci. 2005;360:1617-1638.
3. Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364:1144-1153.
4. Wu L, Williams PM, Koch WH. Clinical applications of microarray-based diagnostic tests. Biotechniques. 2005;39:577-582.
5. Albertini L, Siest G, Jeannesson E, Visvikis-Siest S. Availability of pharmacogenetic and pharmacogenomic information in anticancer drug monographs in France: personalized cancer therapy. Pharmacogenomics. 2011;12:681-691.
6. Yan L, Beckman RA. Pharmacogenetics and pharmacogenomics in oncology therapeutic antibody development. Biotechniques. 2005;39:565-568.
7. Phillips KA, Van Bebber SL. Regulatory perspectives on pharmacogenomics: a review of the literature on key issues faced by the U.S. Food and Drug Administration. Med Care Res Rev. 2006;63:301-326.
8. Workman P. The opportunities and challenges of personalized genome-based molecular therapies for cancer: targets, technologies, and molecular chaperones. Cancer Chemother Pharmacol. 2003;52(suppl 1):S45-S56.
9. Table of pharmacogenomic biomarkers in drug labels. FDA.
www.fda.gov/drugs/
10. Tesch G, Amur S, Schousboe JT, et al. Successes achieved and challenges ahead in translating biomarkers into clinical applications. AAPS J. 2010;12:243-253.
11. Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1-22.
12. Siena S, Sartore-Bianchi A, Di Nicolantonio F, et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308-1324.
13. Vlahovic G, Crawford J. Activation of tyrosine kinases in cancer. Oncologist. 2003;8:531-538.
14. Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040-2048.
15. Hecht JR, Patnaik A, Berlin J, et al. Panitumumab monotherapy in patients with previously treated metastatic colorectal cancer. Cancer. 2007;110:980-988.
16. Pérez-Soler R, Chachoua A, Hammond LA, et al. Determinants of tumor response and survival with erlotinib in patients with non–small-cell lung cancer. J Clin Oncol. 2004;22:3238-3247.
17. Parra HS, Cavina R, Latteri F, et al. Analysis of epidermal growth factor receptor expression as a predictive factor for response to gefitinib (‘Iressa’, ZD1839) in non-small-cell lung cancer. Br J Cancer. 2004;91:208-212.
18. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-1500.
19. Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366-377.
20. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353;133-144.
21. Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102-6114.
22. Diaz NM. Laboratory testing for HER2/neu in breast carcinoma: an evolving strategy to predict response to targeted therapy. Cancer Control. 2001;8:415-418.
23. Blackwell KL, Burstein HJ, Storniolo AM, et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28:1124-1130.
24. NCCN guidelines for patients: breast cancer (v.2.2011). National Comprehensive Cancer Network.
www.nccn.com/images/patient-
25. Van Krieken JH, Jung A, Kirchner T, et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453:417-431.
26. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626-1634.
27. Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408-1417.
28. Erbitux (cetuximab) package insert. Princeton, NJ: Bristol-Myers Squibb; July 2009.
www.accessdata.fda.gov/
29. Vectibix (panitumumab) package insert. Thousand Oaks, CA: Amgen Inc; July 2009.
www.accessdata.fda.gov/
30. Di Bacco AD, Keeshan K, McKenna SL, Cotter TG. Molecular abnormalities in chronic myeloid leukemia: deregulation of cell growth and apoptosis. Oncologist. 2000;5:405-415.
31. Mohamed AN, Pemberton P, Zonder J, Schiffer CA. The effect of imatinib mesylate on patients with Philadelphia chromosome-positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin Cancer Res. 2003;9:1333-1337.
32. Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome–positive ALL. N Engl J Med. 2006;354:2542-2551.
33. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome–positive leukemias. N Engl J Med. 2006;354:2531-2541.
34. Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594-2596.
35. Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360-5367.
36. McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol. 2009;27:5650-5659.
37. Gocek E, Marcinkowska E. Differentiation therapy of acute myeloid leukemia. Cancers. 2011;3:2402-2420.
38. Ravandi F, Estey E, Jones D, et al. Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol. 2009;27:504-510.
39. Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology Provisional Clinical Opinion: Epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non–small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol. 2011;29:2121-2127.
40. Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118-145.
41. Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091-2096.
42. NCCN clinical practice guidelines in oncology: chronic myelogenous leukemia (v.2.2012). National Comprehensive Cancer Network.
www.nccn.org/professionals/
43. Demetri GD, Mehren MV, Antonescu CR, et al. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw. 2010;8(suppl 2):S1-S40.
44. NCCN clinical practice guidelines in oncology: acute myeloid leukemia (v.2.2011). National Comprehensive Cancer Network.
www.nccn.org/professionals/
To comment on this article, contact rdavidson@uspharmacist.com.


