US Pharm. 2013;38(10)(Diabetes suppl):7-10.
ABSTRACT: A common and serious comorbidity in patients with cystic fibrosis (CF) is CF-related diabetes (CFRD), which is caused by mutations in the CFTR gene. The onset of CFRD is insidious and emerges by late adolescence. CFRD significantly increases the mortality rate, so early diagnosis and prompt medical intervention are necessary. The likelihood of microvascular complications increases in patients who have had CFRD for more than 10 years. The main therapy for CFRD is insulin, which controls blood glucose, stabilizes lung function, improves nutritional status, and promotes growth in adolescents. Oral hypoglycemic agents are contraindicated because of adverse effects. Nutritional support is important in the management of CFRD. The pharmacist can advise the patient about medications, dosages, and harmful drug-drug interactions.
Cystic fibrosis (CF) is the most common life-threatening autosomal-recessive genetic condition, affecting 1 in every 3,500 births in the United States.1 This disease is caused by mutations in the CFTR gene, the protein product of which is a cAMP-regulated chloride ion channel that is essential for regulating the movement of salt and water across epithelial cell membranes.2 These mutations affect all exocrine glands, leading to thick mucus that obstructs the glandular ducts and conducting airways.3 Although CF is fatal, advances in medical intervention have made early detection and diagnosis possible, thereby dramatically increasing the survival rate. The median survival age has risen from 1 year in 1950 to 38.3 years in 2010. Despite their increased life expectancy, CF patients today face new complications.4 One of the most common comorbidities in CF patients is CF-related diabetes (CFRD), which further complicates the CF and leads to poorer nutritional status, worsened lung function, and increased mortality.5
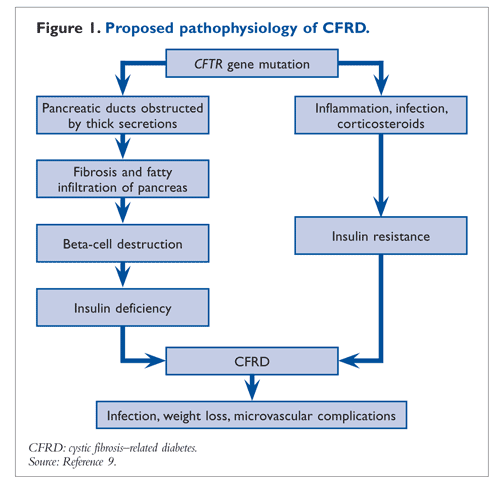
CFRD is usually diagnosed by late adolescence or early adulthood. The average age of onset is 18 to 21 years, and the incidence is slightly higher in females.6 The prevalence of CFRD in adolescents is much higher (19%) than in children younger than 10 years (2%), and the prevalence continues to increase with age.7 CF patients with liver disease are at higher risk for developing CFRD.8 Additionally, the use of oral corticosteroids during acute illness increases a patient’s susceptibility to developing CFRD. The other, less prominent, cause of CFRD is insulin resistance from acute respiratory infections and inflammation (FIGURE 1).9
Prevalence and Symptoms
The prevalence of CFRD has increased dramatically, from 1% in 1962 to 31% in 2007.10 The incidence of CFRD rises with age (40% in patients aged 20-29 years and 50% in those aged 30-39 years). According to a recent report, 30,000 people in the U.S. and 70,000 worldwide have CF.1,11 About 10 million Americans are estimated to carry this defective gene, and 1,000 new cases are diagnosed annually. Between 1999 and 2006, 3,708 CF-related deaths were reported.12
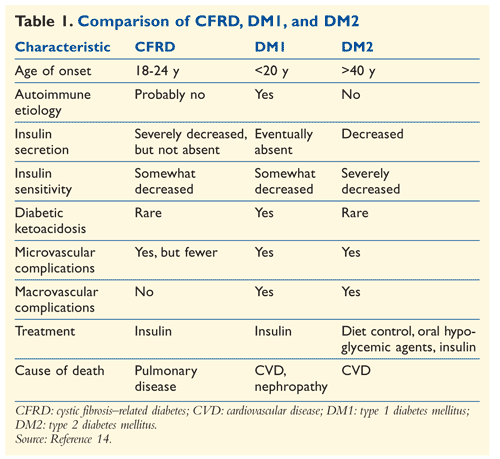
Although some features of CFRD are similar to those of type 1 and type 2 diabetes mellitus (DM1 and DM2, respectively), CFRD is a distinct clinical entity (TABLE 1). CFRD is characterized by the following symptoms: unexplained polyuria or polydipsia; trouble maintaining weight despite nutritional intervention, or failure to gain weight; low growth rate; delayed pubertal progression; and unexplained chronic decline in pulmonary function.13
Pathophysiology and Prognosis
The cause of CFRD is not fully understood, although some scientists believe it to be the destruction of pancreatic islets as a result of fibrosis and fatty infiltration (FIGURE 1). All CF patients with or without CFRD, but with insufficient exocrine function, manifest some degree of beta-cell dysfunction.14 Abnormal chloride channel function in CF results in thick, viscous secretions that obstruct the pancreatic ducts, leading to interstitial edema, ischemic damage of the exocrine pancreas, and beta-cell death.15 The loss of beta cells leads to insulin deficiency, which becomes the primary cause of CFRD.5 One study implicated serum antibodies formed against some bacterial infections in the development of CFRD.16
A retrospective analysis demonstrated higher survival at 30 years in CF patients without CFRD than in those with CFRD (60% vs. 25%).17 The mortality rate is six times higher in CF patients with CFRD than in those without it.13 The incidence of CFRD-related microvascular complications such as retinopathy (16%), microalbuminuria (14%), autonomic neuropathy (52%), and nephropathy is increasing in patients who have had CFRD for more than 10 years.18 Evidence of macrovascular complications has not been reported to date, but if the survival age continues to increase beyond a certain limit, cardiovascular complications may emerge.5 Compared with the adverse effects (AEs) of hyperglycemia, the absence of anabolic effects due to the reduced insulin in CFRD may have serious consequences, including protein catabolism and loss of muscle mass.14 Increased protein catabolism from insulin deficiency is reported to contribute to higher mortality in CFRD.19
Screening and Diagnosis
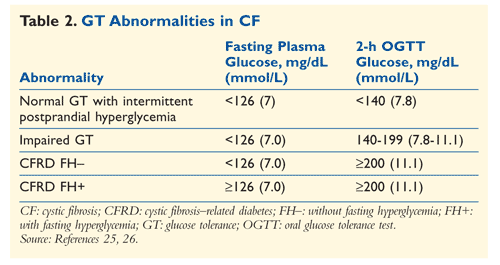
Although glycosylated hemoglobin (A1C), fructosamine, urine glucose, and random glucose levels are used to diagnose DM1 and DM2, these tests are not recommended for diagnosis of CFRD.20-23 The oral glucose tolerance test (OGTT) is the standard test for CFRD, although continuous glucose monitoring could detect abnormal glucose tolerance earlier than OGTT and is validated for use in children and adolescents.14 A CF patient should be screened for CFRD with 2-hour, 75-g OGTT when he or she reaches 10 years of age. The American Diabetes Association’s diagnostic criteria for CFRD are a 2-hour OGTT plasma glucose level of ≥200 mg/dL; a fasting plasma glucose level of ≥126 mg/dL; an A1C ≥6.5%; or polyuria or polydipsia with a casual glucose level >200 mg/dL.24 Patients should be screened annually. In addition, several situations, such as acute illness, continuous-drip enteral feeding, and organ transplantation, also call for CFRD screening (TABLE 2).24-26
Treatment
It is essential that the treatment goals of CFRD include not only hyperglycemia control, reduction of diabetes-associated complications, and avoidance of hypoglycemia, but also maintenance of an optimal nutritional status to achieve normal growth and development in children and adolescents.13 Lack of adherence to the treatment regimen, particularly in adolescents, may result in unnecessary hospitalizations and complications.27 It is extremely important to promote the psychological, social, and emotional adjustment of children and adolescents with chronic illnesses like CFRD.13 Many hypoglycemic drugs are contraindicated because of gastrointestinal (GI) and liver AEs, and they are less effective than insulin. These agents also put an additional load on the beta cells, potentially accelerating CFRD progression.24
Insulin Therapy: Insulin is a standard medical treatment, since CFRD patients are essentially insulin deficient (TABLE 3).24 Many reports indicate that insulin therapy stabilizes lung functions, improves the nutritional status of CFRD patients, improves blood glucose/A1C control, reduces rates of pulmonary exacerbations, and decreases mortality.7,24,26,28-30

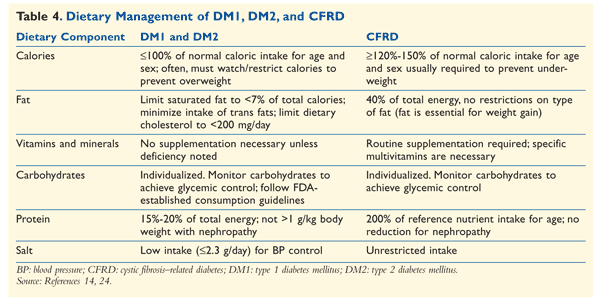
Nutritional Support and Management: Nutritional support is an integral part of CFRD management. An individualized nutrition plan must be developed by the CF specialist dietitian. High-carbohydrate foods and drinks are not restricted in CFRD.31 Dietary requirements are much higher than those for healthy individuals of the same age, sex, and BMI (TABLE 4).32 To maintain growth status, the goal for patients aged 2 to 20 years is a BMI ≥50th percentile. For adults older than 20 years, for optimal growth and sufficient nutritional value, a BMI ≥22 kg/m2 for females and ≥23 kg/m2 for males is recommended.33
Of fats, carbohydrates, and proteins, carbohydrates have the greatest impact on blood sugar levels. It is important to eat carbohydrates at times when there is enough insulin in the body to change them into fuel. CFRD patients on a fixed insulin dosage are better able to manage blood sugar by eating the same amount of carbohydrates during each of their daily three meals and three snacks at about the same time each day. Carbohydrate counting is a method of calculating grams of carbohydrates consumed in meals and snacks. The insulin dose should be matched to the carbohydrate content of the meal at a dose of 0.5 unit/15 g carbohydrate. For proper carbohydrate counting, meals must be planned, foods containing carbohydrates must be identified, and the foods’ carbohydrate content must be calculated.34
Malabsorption of fats and fat-soluble vitamins is highly likely in patients with CFRD.35 Therefore, fat-soluble vitamins (vitamins A, D, E, K), along with minerals such as calcium, iron, sodium, and zinc, are essential. Plasma levels should be checked annually for deficiency, and appropriate supplementation should be administered.36
A multidisciplinary team of health professionals is essential for successful management of CFRD. Among health professionals, a clinical consultant in the field of pharmacology is extremely important. Health-related complications are compounded in CFRD patients, requiring even more constant attention and collaboration with health professionals because of the multiorgan system effects of diabetes in CF. The pharmacist, in particular, can be extremely helpful in advising the patient about medications, dosages, and harmful drug-drug interactions, as well as in assisting in research and recommending appropriate drugs on an individual basis. While no two cases are the same, a multifaceted approach is best for the sustenance of CFRD patients.
Conclusion
CFRD, a common comorbidity in adolescents and adults with CF, develops as a result of insulin deficiency due to a loss of functional pancreatic beta cells and the emergence of insulin resistance through infection, inflammation, and corticosteroid treatment. CFRD is associated with poorer nutritional status, worsened lung function, and increased mortality, thus adding additional treatment and management to an already-debilitating condition. The standard treatment is insulin therapy. Insulin not only controls the hyperglycemia effectively, but also improves nutritional status and significantly reduces lung exacerbation. The use of hypoglycemic agents in the treatment of CFRD is contraindicated because of their GI-system and liver AEs. Nutritional support is extremely important in the management of CFRD, and a nutrition plan tailored to the patient must be developed by the CF specialist dietitian. In addition, psychological, social, and emotional support should be extended to help patients cope with the burdens of CFRD. As life expectancy increases, new complications may surface, so future research must focus on treatment aimed at improving quality of life in patients with CFRD.
REFERENCES
1. Cystic Fibrosis Foundation Patient Registry: Annual Data Report 2010. Bethesda, MD: Cystic Fibrosis Foundation; 2011.
2. Human Genome Project. CFTR: the gene associated with cystic
fibrosis.
www.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/cftr.shtml.
Accessed October 20, 2012.
3. Milla CE. Cystic fibrosis related diabetes. Curr Medical Lit—Diabetes. 2006;23:33-38.
4. Cystic Fibrosis Foundation. Fact sheet.
www.cff.org/UploadedFiles/Chapters/losangeles/ChapterEvents/2010%20Fact%20Sheet.pdf.
Accessed October 15, 2012.
5. Lek N, Acerini CL. Cystic fibrosis related diabetes mellitus—diagnostic and management challenges. Curr Diabetes Rev. 2010;6:9-16.
6. Yung B, Hodson ME. Diabetes in cystic fibrosis. J R Soc Med. 1999;92(suppl 37):35-40.
7. Moran A, Dunitz J, Nathan B, et al. Cystic fibrosis-related
diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32:1626-1631.
8. Marshall BC, Butler SM, Stoddard M, et al. Epidemiology of cystic fibrosis-related diabetes. J Pediatr. 2005;146:681-687.
9. Costa M, Potvin S, Berthiaume Y, et al. Diabetes: a major co-morbidity of cystic fibrosis. Diabetes Metab. 2005;31:221-232.
10. van den Berg JM, Kouwenberg JM, Heijerman HG. Demographics of glucose metabolism in cystic fibrosis. J Cyst Fibros. 2009;8:276-279.
11. Cystic Fibrosis Foundation. About cystic fibrosis: what you need to know. www.cff.org/AboutCF. Accessed October 15, 2012.
12. Cystic fibrosis (CF). In: American Lung Association. State of Lung Disease in Diverse Communities 2010. Washington, DC: National Lung Association; 2010.
13. Moran A, Hardin D, Rodman D, et al. Diagnosis, screening and
management of cystic fibrosis related diabetes mellitus: a consensus
conference report. Diabetes Res Clin Pract. 1999;45:61-73.
14. O’Riordan SM, Robinson PD, Donaghue KC, Moran A. Management of cystic fibrosis-related diabetes. Pediatr Diabetes. 2008;9:338-344.
15. Rana M, Munns CF, Selvadurai H, et al. Cystic fibrosis-related diabetes in children—gaps in the evidence?: key points. Medscape Multispecialty. www.medscape.org/viewarticle/721827_8. Accessed November 10, 2012.
16. Jensen P, Johansen HK, Lanng S, Hoiby N. Relative increase in IgG antibodies to Pseudomonas aeruginosa 60-kDa GroEl in prediabetic patients with cystic fibrosis. Pediatr Res. 2001;49:423-428.
17. Finkelstein SM, Wielinski CL, Elliott GR, et al. Diabetes mellitus associated with cystic fibrosis. J Pediatr. 1988;112:373-377.
18. Schwarzenberg SJ, Thomas W, Olsen TW, et al. Microvascular complications in cystic fibrosis-related diabetes. Diabetes Care. 2007;30:1056-1061.
19. Hardin DS, LeBlanc A, Lukenbaugh S, et al. Proteolysis associated with insulin resistance in cystic fibrosis. Pediatrics. 1998;101:433-437.
20. Lanng S, Hansen A, Thorsteinsson B, et al. Glucose tolerance in patients with cystic fibrosis: five year prospective study. BMJ. 1995;311:655-659.
21. Elder DA, Wooldridge JL, Dolan LM, D’Alessio DA. Glucose
tolerance, insulin secretion, and insulin sensitivity in children and
adolescents with cystic fibrosis and no prior history of diabetes. J Pediatr. 2007;151:653-658.
22. Godbout A, Hammana I, Potvin S, et al. No relationship between
mean plasma glucose and glycated haemoglobin in patients with cystic
fibrosis-related diabetes. Diabetes Metab. 2008;34:568-573.
23. Yung B, Kemp M, Hooper J, Hodson ME. Diagnosis of cystic fibrosis
related diabetes: a selective approach in performing the oral glucose
tolerance test based on a combination of clinical and biochemical
criteria. Thorax. 1999;54:40-43.
24. Moran A, Brunzell C, Cohen RC, et al. Clinical care guidelines
for cystic fibrosis-related diabetes: a position statement of the
American Diabetes Association and a clinical practice guideline of the
Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society.
Diabetes Care. 2010;33:2697-2708.
25. Ode KL, Frohnert B, Laguna T, et al. Oral glucose tolerance testing in children with cystic fibrosis. Pediatr Diabetes. 2010;11:487-492.
26. Mohan K, Israel KL, Miller H, et al. Long-term effect of insulin treatment in cystic fibrosis-related diabetes. Respiration. 2008;76:181-186.
27. La Greca AM, Schuman WB. Adherence to prescribed medical regimens. In: Roberts MC, ed. Handbook of Pediatric Psychology. 2nd ed. New York, NY: Guilford Press; 1995:55-83.
28. Rolon MA, Benali K, Munck A, et al. Cystic fibrosis–related
diabetes mellitus: clinical impact of prediabetes and effects of insulin
therapy. Acta Paediatr. 2001;90:860-867.
29. Hardin DS, Rice J, Rice M, Rosenblatt R. Use of the insulin pump in treat [sic] cystic fibrosis related diabetes. J Cyst Fibros. 2009;8:174-178.
30. Mozzillo E, Franzese A, Valerio G, et al. One-year glargine
treatment can improve the course of lung disease in children and
adolescents with cystic fibrosis and early glucose derangements. Pediatr Diabetes. 2009;10:162-167.
31. Management of Cystic Fibrosis Related Diabetes Mellitus: Report of the UK Cystic Fibrosis Trust Diabetes Working Group. Bromley, England: Cystic Fibrosis Trust; 2004.
32. Vaisman N, Pencharz PB, Corey M, et al. Energy expenditure of patients with cystic fibrosis. J Pediatr. 1987;111:496-500.
33. Stallings VA, Stark LJ, Robinson KA, et al. Evidence-based
practice recommendations for nutrition-related management of children
and adults with cystic fibrosis and pancreatic insufficiency: results of
a systematic review. J Am Diet Assoc. 2008;108:832-839.
34. Brunzell C, Hardin DS, Moran A, et al. Managing Cystic Fibrosis-Related Diabetes “(CFRD)”: An Instruction Guide for Patients and Families. 4th ed. Bethesda, MD: Cystic Fibrosis Foundation; 2008.
35. Cystic Fibrosis Medicine. Vitamins. www.cfmedicine.com/htmldocs/CFText/vitamins.htm. Accessed August 22, 2013.
36. Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35:246-259.
To comment on this article, contact rdavidson@uspharmacist.com.


