US Pharm. 2014;39(5)(Specialty&Oncology suppl):3-8.
ABSTRACT: Pituitary carcinomas, which are extremely rare, are characterized by poor prognosis. Diagnosis is based on the presence of metastases, at which point the tumor becomes unresponsive to therapy. Although various markers of molecular pathogenesis have been proposed, none are reliable predictors of disease progression or outcome. Most pituitary carcinomas are hormonally active, usually hypersecreting adrenocorticotropic hormone or prolactin. Medical interventions for pituitary carcinomas include surgery, radiotherapy, and pharmacotherapy. The paucity of data on pituitary carcinomas renders necessary further research into underlying mechanisms and novel molecular targets for treatment.
Tumors of the pituitary gland are relatively common in the general population, representing 10% to 20% of primary intracranial neoplasms.1,2 According to the 2004 World Health Organization classification of endocrine tumors, pituitary tumors are classified into benign adenomas, atypical (invasive) adenomas, and carcinomas.3 Benign and invasive adenomas constitute the majority of pituitary neoplasms, with an estimated prevalence of approximately 17% and 35%, respectively.4,5 Malignant transformation is quite rare in pituitary adenomas; carcinomas represent 0.1% to 0.2% of all pituitary tumors.6-8 One study reported approximately 165 cases of pituitary carcinomas documented in English-language literature as of 2011, but this may be an underestimation, owing to difficulties in diagnosis and treatment.9-11
Pituitary carcinomas present a challenge to clinical practice. Given the rarity of these carcinomas, no gender- or age-related trends are evident.8 The majority of cases (88%) are hormonally active, with manifestations indistinguishable from those of adenomas (e.g., Cushing disease, hyperprolactinemia, acromegaly).12,13 Prognosis is poor (mean 2 years, range 3 months to 8 years), as these carcinomas are unresponsive to conventional therapies used for endocrinologically active adenomas.1 Treatment strategies include surgery, radiotherapy (RT), and/or pharmacologic agents. This review discusses clinical evaluation and therapies for this uncommon, but significant, disease.PATHOPHYSIOLOGY
Pituitary carcinomas are defined by the presence of cerebrospinal and/or systemic metastasis.3 Whether carcinomas occur de novo or progress from atypical adenomas is unknown.1 Pituitary carcinomas without evidence of a prior benign lesion have been reported.14,15 However, most pituitary carcinomas are believed to arise from aggressive pituitary adenomas that transition to malignancy, leading to development of distant metastases. Supporting evidence comes from 1) initial presentation as macroadenoma (>1 cm), with multiple recurrences despite medical therapy, surgery, and RT16; 2) long latency period for progression to carcinoma (up to 18 years)6; and 3) progressive accumulation of genetic changes.17,18 The most common sites of metastasis are the brain, spinal cord, and meninges.19 Other sites include the bones, liver, lymph nodes, ovaries, heart, and lungs.7
CLINICAL PRESENTATION
Patients with pituitary carcinomas tend to present with a combination of signs and symptoms associated with mass effect and/or excess endocrine function.19,20 Tumor enlargement may result in visual-field defects from direct compression of the optic apparatus or in headaches from elevated intracranial pressure.12,19 Nonfunctioning carcinomas present with mass effect only.20,21 Most functioning carcinomas result in hypersecretion of adrenocorticotropic hormone (ACTH; 42%) and prolactin (33%).7 Less commonly, carcinomas that produce growth hormone (GH; 6%), follicle-stimulating hormone or luteinizing hormone (5%), or thyroid-stimulating hormone (TSH; 1%) have been reported.6,7 Most ACTH-producing tumors manifest as Cushing syndrome.22 Prolactin-secreting tumors present as hypogonadism, infertility, and galactorrhea.23 GH-secreting pituitary carcinomas present as invasive macroadenomas and acromegaly.24
DIAGNOSIS
In most cases, the clinical course of pituitary carcinoma is not distinct from that of pituitary adenoma. Laboratory test results of highly elevated hormone levels despite adequate surgical clearance of the tumor may indicate the presence of metastasis.16 The immunohistochemical profile of the primary tumor and the metastatic deposit may be evaluated at the time of transformation to a carcinoma.19 Typical histologic features of malignancy include hypercellularity, pleomorphism, mitotic activity, necrosis, and invasion of surrounding structures.1,13 The cell cycle–specific nuclear antigen Ki-67 has been shown to be increased, and a labeling index threshold of 3% has been proposed to differentiate invasive and noninvasive tumors (97% specificity, 73% sensitivity).25 Molecular oncogenes such as p53, p27, and multiple endocrine neoplasia type 1 have been found to be altered, but mutations that fully explain carcinoma tumorigenesis remain elusive.1,8,17,19,26-28
Given the lack of diagnostic biomarkers, multiple recurrences requiring repeated medical interventions may be noted before metastasis is detected and definitively diagnosed.13 A differential to evaluate is the presence of a primary distant tumor site with metastasis to the pituitary versus a primary pituitary carcinoma. Distant metastasis is necessary for a diagnosis of pituitary carcinoma. High-resolution gadolinium-enhanced MRI is the preferred diagnostic imaging tool for evaluating metastatic disease.13,29 However, neuroimaging does not differentiate locally invasive pituitary adenomas from pituitary tumors that will progress to carcinoma.13
TREATMENT
Once a pituitary carcinoma manifests as systemic and/or craniospinal metastases, therapeutic options are limited. In some cases, the patient dies from complications of hormonal excess (e.g., immunosuppression, dysglycemia, poor wound healing) before mass effects from the expanding tumor. The same principles applied to the treatment of benign pituitary tumors are applied to carcinomas. Treatment modalities include surgical resection, RT, and pharmacotherapy; however, no randomized trials have compared these strategies. Treatment of pituitary carcinomas is palliative.30 Goals of therapy are symptom relief and maintenance of quality of life.
Nonpharmacologic Options
Surgery: Pituitary carcinomas typically are large (>1 cm). Although first-line therapy involves surgical resection, surgical procedures are rarely curative, possibly because of the invasive nature or incomplete resection of the tumor.16 Tumor recurrence seems to be highest in patients with a prolactin-secreting carcinoma.31 Recurrence usually takes place 1 to 5 years after surgery.31 Transsphenoidal endoscopic or transcranial surgery is combined with other treatment modalities, and repeat surgeries may be necessary for metastatic control and for decompression of vital central nervous system (CNS) structures.9,31
RT/Radiosurgery: RT has been utilized as an adjunct to surgical measures, to prevent tumor regrowth, and to slow the spread of expanding tumors and/or metastases.9,32 RT may benefit patients who cannot tolerate surgery or those in whom surgical excision is not possible. Although improved survival has not been shown in clinical trials, a few case reports indicate delayed tumor progression.9 Methods include Gamma Knife, CyberKnife, linear accelerator, and proton beam therapy administered according to one of two main schedules—stereotactic radiosurgery (SRS) or fractionated RT.2,33 SRS usually involves delivery of high-dose RT in a single visit, whereas fractionated RT consists of smaller daily-dose fractions given over 5 to 6 weeks. Adverse effects (AEs) include long-term hypopituitarism and necrosis of certain brain areas.34
Pharmacologic Options
Pharmacotherapy involves agents aimed primarily at controlling biochemical hypersecretion and proliferation to reduce hormone production and slow tumor growth (TABLE 1). Treatments for hormone-level reduction are similar to those employed for pituitary adenomas, although higher doses and combinations of agents are needed.9 Given the rarity of pituitary carcinomas, no randomized studies of systemic chemotherapy (ChT) have been conducted.9 Many single-agent and combination ChT regimens have been used, with variable response rates.35
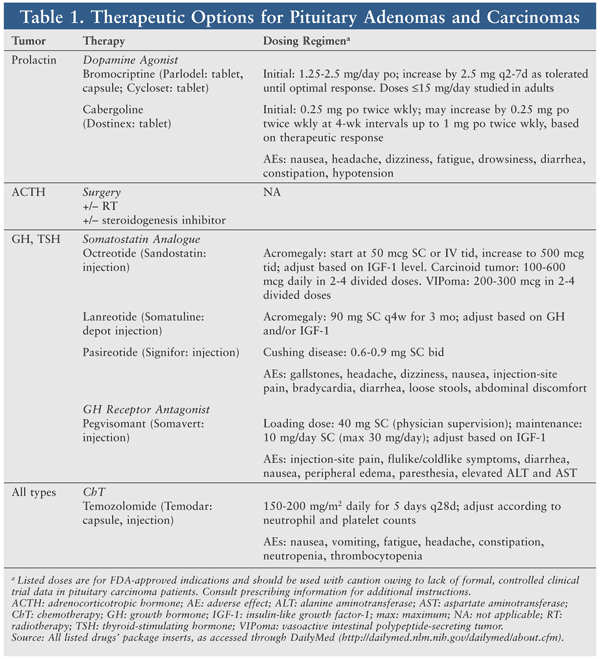
Hormone-Targeted Therapy
Dopamine Agonists: Dopamine agonists (DAs) activate postsynaptic dopamine receptors. The release of dopamine from tuberoinfundibular neurons in the hypothalamus modulates prolactin secretion by the anterior pituitary gland.23 DAs significantly reduce plasma levels of prolactin in patients with physiologically elevated prolactin and in those with hyperprolactinemia. Bromocriptine (Parlodel, Cycloset) and cabergoline (Dostinex) have been used to control prolactinoma size and hypersecretion, with an overall success rate of 80% to 95%.1,12 Unfortunately, these agents are palliative only in the case of metastatic prolactin-secreting carcinomas.36
DAs are typically administered at a higher dose, which may elicit a temporary therapeutic response and lead to AEs (e.g., nausea, hypotension). However, the carcinoma may become resistant to, or “escape,” the DA’s effects.6 The serum prolactin level may reach up to 600 times the upper limit of normal.7 A sudden, marked elevation of prolactin may reflect tumor recurrence and/or metastasis.
Although optimal management requires further investigation, DAs remain first-line treatment for symptomatic prolactinoma. According to the Endocrine Society’s practice guidelines for hyperprolactinemia, cabergoline is preferred over other DAs because it is more effective at normalizing prolactin levels and is associated with a greater frequency of pituitary tumor shrinkage.23 The greater efficacy may be explained by cabergoline’s higher affinity for dopamine receptor (i.e., D2) binding sites. The incidence of AEs is lower with cabergoline, potentially leading to better tolerance and adherence.
No clinical trials have directly compared the mass-reducing effects of different DAs.23,37 Given estrogen receptor expression on some prolactin-secreting tumors, antiestrogens such as tamoxifen have been used (albeit unsuccessfully) to achieve synergism.16 Unresponsive prolactinomas may be treated with surgery or ChT. DAs have been used to treat malignant GH-secreting (cabergoline), ACTH-secreting, and TSH-secreting pituitary carcinomas, with little to no benefit.9
Somatostatin Analogues (SSAs): Somatostatin (SST) is a hormone produced by neurons in the periventricular nucleus of the hypothalamus, as well as other CNS sites. SST exerts its effects on the anterior pituitary gland, binding to SST receptors and preventing release of GH, TSH, gastrointestinal hormones, pancreatic enzymes, and neuropeptides.38 GH overproduction can manifest as acromegaly, characterized by elevated insulin-like growth factor-I (IGF-1) and subsequent somatic growth and metabolic dysfunction.
SSAs such as octreotide (Sandostatin) and lanreotide (Somatuline) have been used to treat GH-secreting pituitary adenomas, as well as those secreting TSH, ACTH, and prolactin.16 SSAs effectively normalize IGF-1 and GH levels in approximately 55% of acromegaly-diagnosed patients.39 SSAs modestly reduce pituitary tumor size in 25% to 70% of patients, depending upon whether they are used as adjuvant or de novo therapy.39 The long-acting, subcutaneously administered formulations of octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot) have similar pharmacologic and efficacy profiles. According to the American Association of Clinical Endocrinologists, the short-acting octreotide formulation is effective and may be used, especially if there are financial constraints or a rapid onset of action is necessary.39 In patients with a partial response to SSAs, adding cabergoline or a GH antagonist may be effective for further lowering GH or IGF-1 levels.39
Therapeutic outcomes of short- and long-acting SSAs for pituitary carcinomas have been variable and largely disappointing.9,16 This may be due partly to different expression levels and affinities for certain SST receptor subtypes (i.e., 2 and 5) or to posttranscriptional effects.16 A next-generation analogue, pasireotide (Signifor), exhibits 40-fold increased affinity for SST subtype 5, providing targeted reduction of plasma ACTH and cortisol in patients with Cushing disease.40 One case of combination pasireotide-temozolomide therapy in a patient with an ACTH-secreting metastasized pituitary carcinoma has been reported.41
Growth Hormone Antagonist: Pegvisomant (Somavert) is a GH receptor antagonist that selectively binds to GH receptors on cell surfaces and blocks binding of endogenous GH, consequently interfering with GH signal transduction and decreasing IGF-1 production.42 Pegvisomant is indicated to treat refractory acromegaly and has been used (alone or with SSAs) to treat GH-secreting adenomas.16,39 Pegvisomant is highly effective at normalizing IGF-1 values (>90%) in acromegaly patients who are partially or completely resistant to other therapies.39 In patients with partial response to SSA therapy, the addition of daily, weekly, or twice-weekly pegvisomant may be beneficial.39 However, use in patients with malignant GH-secreting tumors has not been well studied.
Other: As previously mentioned, ACTH overproduction can present as Cushing syndrome. Although the primary treatment is surgery, drugs that suppress cortisol hypersecretion may be prescribed in cases in which surgery is unsuccessful or contraindicated. Ketoconazole acts as an inhibitor of steroidogenesis in the adrenal glands. Although effective for recurrent Cushing syndrome, chronic ketoconazole treatment is limited by AEs (e.g., hepatotoxicity).43 Ketoconazole for pituitary carcinoma treatment requires further study.
Chemotherapy
Temozolomide: Few cases of short-term stabilization with systemic ChT have been reported. However, ChT should be an option for pituitary carcinomas previously treated with both surgery and RT. Temozolomide (Temodar), which is indicated for the treatment of glioblastoma multiforme, has demonstrated some efficacy against pituitary carcinomas.44 Temozolomide is administered orally (100% bioavailability), crosses the blood-brain barrier, and is not cell cycle–specific.9 Temozolomide undergoes rapid conversion at physiological pH to the methylating alkylator compound 5-(3-methyltriazen-1-yl)-imidazole-4-carboxamide (MTIC). MTIC induces cell damage by methylating DNA at the O6 and N7 positions of guanine, causing base pair mismatch of O6-methylguanine with thymidine in the sister chromatid instead of cytosine.9 Temozolomide seems effective in all pituitary carcinoma subtypes, but the optimal duration (≥12 cycles?) and dosage for naïve and stabilized, responsive patients are poorly defined. The regimen is generally well tolerated; fatigue and myelosuppression are expected, requiring dose reduction and occasional withdrawal of the agent. The increase in survival (6 months to <2 years) could be considered insignificant, but is undoubtedly invaluable to patients.
Other: Other agents used in ChT protocols, alone or in combination, include lomustine, 5-fluorouracil, carmustine, hydroxyurea, interferon alfa, cisplatin, carboplatin, paclitaxel, etoposide, doxorubicin, dacarbazine, cyclophosphamide, procarbazine, vincristine, mitotane, and methotrexate.7,9 Most combinations of cytotoxic drugs have yielded poor response rates, possibly owing to well-differentiated proliferative phenotypes of pituitary carcinoma cells.9
CONCLUSION
Pituitary carcinomas are rare and present diagnostic and therapeutic challenges. Despite multiple aggressive treatment approaches, therapies are palliative, and 75% of patients with systemic metastasis die within 1 year of documented spread.44 Given the small number of reported cases, many questions regarding optimal therapy remain unanswered. Future disease management requires more evidence in the form of in vitro and clinical studies concerning the molecular mechanisms of pathogenesis and disease progression.
REFERENCES
1. Oh MC, Tihan T, Kunwar S, et al. Clinical management of pituitary carcinomas. Neurosurg Clin N Am. 2012;23:595-606.
2. Mehta GU, Jane JA Jr. Pituitary tumors. Curr Opin Neurol. 2012;25:751-755.
3. DeLellis RA, Lloyd RV, Heitz PU, Eng C, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon, France: IARC Press; 2004.
4. Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613-619.
5. Scheithauer BW, Kovacs KT, Laws ER Jr, Randall RV. Pathology of
invasive pituitary tumors with special reference to functional
classification. J Neurosurg. 1986;65:733-744.
6. Pernicone PJ, Scheithauer BW, Sebo TJ, et al. Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer. 1997;79:804-812.
7. Ragel BT, Couldwell WT. Pituitary carcinoma: a review of the literature. Neurosurg Focus. 2004;16:E7.
8. Scheithauer BW, Kurtkaya-Yapicier O, Kovacs KT, et al. Pituitary carcinoma: a clinicopathological review. Neurosurgery. 2005;56:1066-1074.
9. Heaney AP. Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab. 2011;96:3649-3660.
10. Daly AF, Tichomirowa MA, Beckers A. The epidemiology and genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2009;23:543-554.
11. Kontogeorgos G. Classification and pathology of pituitary tumors. Endocrine. 2005;28:27-35.
12. Colao A, Ochoa AS, Auriemma RS, et al. Pituitary carcinomas. Front Horm Res. 2010;38:94-108.
13. Lopes MB, Scheithauer BW, Schiff D. Pituitary carcinoma: diagnosis and treatment. Endocrine. 2005;28:115-121.
14. Luzi P, Miracco C, Lio R, et al. Endocrine inactive pituitary
carcinoma metastasizing to cervical lymph nodes: a case report. Hum Pathol. 1987;18:90-92.
15. Nudleman KL, Choi B, Kusske JA. Primary pituitary carcinoma: a clinical pathological study. Neurosurgery.1985;16:90-95.
16. Kaltsas GA, Nomikos P, Kontogeorgos G, et al. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. 2005;90:3089-3099.
17. Dworakowska D, Grossman AB. The pathophysiology of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2009;23:525-541.
18. Syro LV, Scheithauer BW, Kovacs K, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics (Sao Paulo). 2012;67(suppl 1):43-48.
19. Sansur CA, Oldfield EH. Pituitary carcinoma. Semin Oncol. 2010;37:591-593.
20. Nemergut EC, Dumont AS, Barry UT, Laws ER. Perioperative
management of patients undergoing transsphenoidal pituitary surgery. Anesth Analg. 2005;101:1170-1181.
21. Korbonits M, Carlsen E. Recent clinical and pathophysiological advances in non-functioning pituitary adenomas. Horm Res. 2009;71(suppl 2):123-130.
22. van der Klaauw AA, Kienitz T, Strasburger CJ, et al. Malignant
pituitary corticotroph adenomas: report of two cases and a comprehensive
review of the literature. Pituitary. 2009;12:57-69.
23. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and
treatment of hyperprolactinemia: an Endocrine Society clinical practice
guideline. J Clin Endocrinol Metab. 2011;96:273-288.
24. Kiseljak-Vassiliades K, Shafi S, Kerr JM, et al. Clinical implications of growth hormone-secreting tumor subtypes. Endocrine. 2012;42:18-28.
25. Thapar K, Kovacs K, Scheithauer BW, et al. Proliferative activity
and invasiveness among pituitary adenomas and carcinomas: an analysis
using the MIB-1 antibody. Neurosurgery. 1996;38:99-106.
26. Salehi F, Agur A, Scheithauer BW, et al. Ki-67 in pituitary neoplasms: a review—part I. Neurosurgery. 2009;65:429-437.
27. Rostad S. Pituitary adenoma pathogenesis: an update. Curr Opin Endocrinol Diabetes Obes. 2012;19:322-327.
28. Gadelha MR, Trivellin G, Hernández Ramírez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res. 2013;41:111-140.
29. Thapar K, Laws ER Jr. Pituitary tumors. In: Kaye AH, Laws ER Jr, eds. Brain Tumors: An Encyclopedic Approach. 2nd ed. London, England: Churchill Livingstone; 2001.
30. Raverot G, Castinetti F, Jouanneau E, et al. Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment. Clin Endocrinol (Oxf). 2012;76:769-775.
31. Roelfsema F, Biermasz NR, Pereira AM. Clinical factors involved
in the recurrence of pituitary adenomas after surgical remission: a
structured review and meta-analysis. Pituitary. 2012;15:71-83.
32. Platta CS, Mackay C, Welsh JS. Pituitary adenoma: a radiotherapeutic perspective. Am J Clin Oncol. 2010;33:408-419.
33. Jagannathan J, Yen CP, Pouratian N, et al. Stereotactic
radiosurgery for pituitary adenomas: a comprehensive review of
indications, techniques and long-term results using the Gamma Knife. J Neurooncol. 2009;92:345-356.
34. Winder MJ, Mayberg MR. Recent advances in pituitary tumor management. Curr Opin Endocrinol Diabetes Obes. 2011;18:278-288.
35. Komninos J, Vlassopoulou V, Protopapa D, et al. Tumors metastatic
to the pituitary gland: case report and literature review. J Clin Endocrinol Metab. 2004;89:574-580.
36. Winkelmann J, Pagotto U, Theodoropoulou M, et al. Retention of
dopamine 2 receptor mRNA and absence of the protein in craniospinal and
extracranial metastasis of a malignant prolactinoma: a case report. Eur J Endocrinol. 2002;146:81-88.
37. dos Santos Nunes V, El Dib R, Boguszewski CL, Nogueira CR.
Cabergoline versus bromocriptine in the treatment of hyperprolactinemia:
a systematic review of randomized controlled trials and meta-analysis. Pituitary. 2011;14:259-265.
38. Anthony L, Freda PU. From somatostatin to octreotide LAR: evolution of a somatostatin analogue. Curr Med Res Opin. 2009;25:2989-2999.
39. Katznelson L, Atkinson JL, Cook DM, et al. American Association
of Clinical Endocrinologists medical guidelines for clinical practice
for the diagnosis and treatment of acromegaly—2011 update. Endocr Pract. 2011;17(suppl 4):1-44.
40. Colao A, Faggiano A, Pivonello R. Somatostatin analogues: treatment of pituitary and neuroendocrine tumors. Prog Brain Res. 2010;182:281-294.
41. Bode H, Seiz M, Lammert A, et al. SOM230 (pasireotide) and
temozolomide achieve sustained control of tumour progression and ACTH
secretion in pituitary carcinoma with widespread metastases. Exp Clin Endocrinol Diabetes. 2010;118:760-763.
42. Sherlock M, Woods C, Sheppard MC. Medical therapy in acromegaly. Nat Rev Endocrinol. 2011;7:291-300.
43. Fleseriu M, Petersenn S. Medical management of Cushing’s disease: what is the future? Pituitary. 2012;15:330-341.
44. Ortiz LD, Syro LV, Scheithauer BW, et al. Temozolomide in aggressive pituitary adenomas and carcinomas. Clinics (Sao Paulo). 2012;67(suppl 1):119-123.
To comment on this article, contact rdavidson@uspharmacist.com.


