ABSTRACT: Angioedema (AE) is localized, nonpruritic swelling resulting from a transient increase in the permeability of postcapillary venules of subcutaneous and submucosal tissue. The swelling can occur at any location and may last between 2 and 5 days. Common locations of swelling include the face, extremities, and gastrointestinal tract, including the bowel wall. Laryngeal edema, while less common, is the most serious presentation because of the high rate of associated mortality. The various syndromes of AE include hereditary, acquired, idiopathic, and drug-induced. Triggers of, and treatments for, the various syndromes may differ; therefore, it is important to understand which type of AE a patient presents with.
US Pharm. 2013;38(6):HS2-HS5.
Angioedema (AE), a disease involving self-limited, localized, nonpruritic swelling, occurs in approximately 15% of the population.1-3 In AE, the edema is caused by the release of vasoactive mediators and a transient increase in the permeability of postcapillary venules of subcutaneous and submucosal tissues.2 Urticaria may occur in 50% of AE cases.1,2
A swelling episode often lasts between 2 and 5 days.1,2 Swelling can occur at any location; however, common sites include the face (periorbital area, lips, tongue), extremities, and gastrointestinal tract or bowel wall.1-3 Laryngeal edema, the most serious presentation, is associated with a mortality rate of 25% to 40%.2
Several different syndromes of AE exist: hereditary (HAE), acquired (AAE), idiopathic (IAE), and drug-induced (DAE). AE may be caused by other syndromes or triggers; however, these factors are beyond the scope of this article. HAE and AAE are considered to be chronic syndromes and do not present with urticaria.1 These conditions are generally mediated by bradykinin and thus are unresponsive to antihistamine therapy. By comparison, AE associated with allergic reactions and urticaria often is responsive to antihistamine therapy. IAE is a diagnosis of exclusion and represents a varied spectrum of disease.
THE AE SYNDROMES
Hereditary
HAE is an autosomal-dominant disorder characterized by recurrent nonpruritic edema of the skin and submucosal tissues.1,2,4-6 The prevalence of HAE ranges from 1 in 10,000 to 1 in 50,000 persons in the United States.4,7 Prevalence is not affected by sex or ethnicity; however, women may have more severe disease.1,4,7 A family history is present in approximately 75% of cases, indicating genetic inheritance; however, 25% of cases are thought to be due to a spontaneous mutation (i.e, a family history is absent).7 Patients often experience disease onset and a swelling episode during childhood, with an increase in severity during puberty.4,5,7,8 The frequency of attacks, which varies between patients, may be weekly or yearly.8
HAE is a congenital quantitative or functional deficiency of C1 esterase inhibitor (C1-INH); it is not associated with a hypersensitivity to foods or other allergens.1,4,7 C1-INH regulates the activation of the complement and contact systems and is involved in the regulation of fibrinolysis and coagulation.1 A low level of C1-INH or poorly functioning C1-INH allows unopposed activation of the complement system via C1. C1-INH inactivates factor XIIa, plasmin, and kallikrein, which feature prominently in the production of bradykinin.4 Thus, the lack of inhibition by C1-INH leads to unregulated synthesis of bradykinin and complement fragments. Excess production of bradykinin, a potent vasodilatory peptide, is responsible for the edema in HAE patients.4,5,7,8
Three types of HAE are documented. Type 1 is the most common and is responsible for approximately 85% of HAE cases.7,8 Patients with type 1 HAE have decreased production of C1-INH, with a reduction in functional activity. Type 2 HAE, which is less common (occurring in approximately 15% of HAE patients), is characterized by dysfunctional C1-INH. In type 2 HAE, C1-INH is measurable at normal or elevated levels. Type 3 HAE is rare and is considered to occur in women, but it has been observed in men as well.6,7 Type 3 HAE is associated with normal C1-INH levels and is believed to be estrogen dependent.2,7,8
In HAE, swelling episodes are generally self-limiting and most often affect the skin, face, extremities, gastrointestinal tract, and upper respiratory tract; occasionally, the laryngeal structures are affected (TABLE 1).1,4,7,8 It is estimated that 50% of HAE patients will experience laryngeal swelling during their lifetime.1,7 Approximately 40% to 87% of HAE episodes are preceded by a prodromal syndrome.9 The prodromal phase may consist of tingling skin, local discomfort, fatigue, or erythema marginatum, a characteristic nonpruritic rash involving the trunk and inner surfaces of the arms and legs.1,4,7 Common triggers include trauma, surgery, menstruation, infection, stress, oral contraceptives, hormone replacement therapy (HRT), and ACE inhibitors. Therefore, patients with HAE should avoid ACE inhibitors, estrogen contraceptives, and HRT.2,6

Treatment of HAE involves the management of acute attacks, as well as prophylactic therapy for some patients. Airway protection should remain a priority in patients with laryngeal involvement.2 Acute treatments are effective for swelling at any site and include plasma-derived C1-INH, plasma kallikrein inhibitor, and selective bradykinin B2-receptor antagonist (TABLE 2).9-15 The use of fresh frozen plasma for acute treatment of HAE is controversial since it also contains kininogen and may increase bradykinin production.1,8 H1-receptor antagonists, glucocorticoids, and epinephrine are not effective for treating HAE.7,8
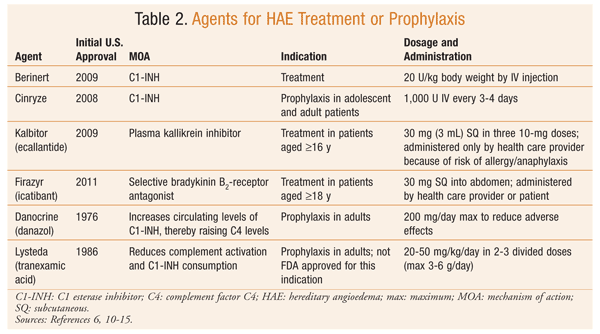
Chronic prophylactic therapy is indicated for specific HAE patients (TABLE 3).2,4,5 Plasma-derived purified C1-INH, approved for such use, is associated with fewer side effects than other therapies indicated for prophylaxis. While plasma-derived C1-INH has been shown to be an effective treatment, prophylaxis does not completely abolish episodes of swelling in all patients.9 One major concern with plasma-derived therapy is viral transmission.

Danazol, an attenuated androgen, also is effective for prophylaxis. Androgens have many adverse effects, and these may limit long-term use, particularly at doses greater than 200 mg (danazol).5,7,9 Adverse effects include virilization, hepatotoxicity, weight gain, menstrual abnormalities, anxiety, and altered libido and mood. CBC, liver enzymes, and lipids should be monitored regularly in patients receiving androgen therapy.5 Antifibrinolytic agents, such as tranexamic acid, may be used in children or adults who have failed or cannot tolerate other therapies.5,7,9 Adverse effects associated with antifibrinolytics include nausea, diarrhea, muscle weakness, hypotension, increased fatigue, enhanced thrombosis, and teratogenicity. Antifibrinolytic agents have not been shown to be as effective as other available agents for treating HAE, and they are less effective for severe or established attacks.9
Short-term prophylaxis is recommended prior to an event that may induce an acute exacerbation.4,8,9 Consensus guidelines recommend the use of C1-INH concentrate prior to a dental or other surgical procedure. Alternatively, attenuated androgens may be used at a doubled dosage for 5 days before and 2 days after the procedure or event. Fresh frozen plasma (2-3 U) also may be useful for short-term prophylaxis.8,9
Acquired
AAE, also known as acquired C1 inhibitor deficiency, results from either increased consumption (type 1) or inactivation (type 2) of C1-INH.1,8 Type 1 AAE usually occurs in patients with lymphoproliferative diseases. Patients have antibodies against immunoglobulins (Ig) on B cells, which combine to create immune complexes. The immune complexes then activate C1. The demands of C1-INH are too great for the large quantities of C1 produced, and AE occurs. Type 2 AAE, which is associated with autoimmune disease, results from autoantibodies to C1-INH.1,2 The autoantibody, usually IgG, binds to and inactivates the C1-INH.8 The inactive fragment is cleaved and yet is detected as normal, thereby rendering quantitative measurements of C1-INH normal. Functional analysis of the C1-INH uncovers its inactivity, however. The role of C1-INH in the pathophysiology of AE is discussed above.
The triggers and clinical characteristics of AAE are similar to those of HAE; however, AAE is differentiated by a lack of family history, a later disease onset (typically fourth decade of life), and serum C1q concentrations (a laboratory parameter that is helpful in differentiating HAE and AAE).1,8 Treatment for an acute onset of swelling in AAE patients is similar to treatment for HAE.5 C1-INH concentrate, ecallantide, and icatibant may be utilized for acute attacks. Higher doses of C1-INH concentrate may be required for AAE patients than for HAE patients, and C1-INH concentrate may become ineffective over time in AAE.3,5 Treatment of the underlying lymphoproliferative disorder should also be addressed.1
Prophylactic therapy for AAE may be required in some cases. As with HAE, options include plasma-derived C1-INH, attenuated androgens, and tranexamic acid. However, in AAE, antifibrinolytics may be more beneficial than attenuated androgens.3,7
Idiopathic
IAE is a diagnosis of exclusion and often is viewed as the most common cause of chronic angioedema; however, the true prevalence is unknown.1,2 The exact mechanisms involved in IAE are not known.5 Based on patient response to various medications, some cases are thought to be mediated by IgE-independent mechanisms that cause mast-cell activation. First-line treatment for patients with IAE without urticaria is antihistamines.3 During acute attacks, a combination of antihistamines, corticosteroids, and epinephrine may be used effectively. If antihistamines are not effective, antifibrinolytics may relieve symptoms. The patient should be observed, and the airway must be protected if necessary. The differential diagnosis for IAE patients should include HAE, AAE, DAE, allergic conditions (including food allergy and latex allergy), Gleich’s syndrome, and idiopathic capillary leak syndrome.1
Drug-Induced
ACE inhibitors are the most common class of drugs associated with AE, with swelling occurring in 0.1% to 6% of treated patients.1,2,5 Patients taking ACE inhibitors who are female, current smokers, older than 65 years, and/or African-American are at increased risk.1,5,16 DAE often occurs during the first month of treatment, but it may present at any time, including years after initiation.16 ACE inhibitor–associated AE frequently causes swelling of the face, tongue, and lips.1
The etiology of ACE inhibitor–induced AE is unclear; however, the edema is believed to be caused by an impaired degradation of peptides, such as bradykinin.7,16 Evidence implicating substance P in DAE also exists.16 Treatment requires discontinuation of the medication. Symptoms may improve in 24 to 48 hours after discontinuation. More severe cases may require intubation or cricothyroidotomy. Corticosteroids and antihistamines are not effective. Icatibant has been used, but it is not FDA approved for this indication.
Other drugs that have been documented to cause AE include angiotensin receptor blockers, aspirin, nonsteroidal anti-inflammatories (NSAIDs), narcotics, antibiotics, interleukin-2, oral contraceptives, and interferon alfa.1,16 Female sex, atopy, and previous allergic reaction may increase the risk of AE from aspirin and NSAIDs.16 The AE episode associated with these drugs may result from an anaphylactic reaction related to antigen-specific IgE, or it may result from an anaphylactoid reaction with IgE involvement.
CONCLUSION
It is important for pharmacists to understand the various syndromes of AE. Treatment may differ based on the pathophysiology of swelling with which the patient presents. When caring for patients with AE, it is important to consider the specific syndrome, the attack severity, and the attack frequency. AE that involves laryngeal swelling is life-threatening and should be considered an emergency. Patients should be counseled regarding specific triggers to avoid, as well as the use of prophylactic therapy when necessary.
REFERENCES
1. Banerji A, Sheffer AL. The spectrum of chronic angioedema. Allergy Asthma Proc. 2009;30:11-16.
2. Temiño VM, Peebles RS Jr. The spectrum and treatment of angioedema. Am J Med. 2008;181:282-286.
3. Grigoriadou S, Longhurst HJ. Clinical immunology review series. An approach to the patient with angio-oedema. Clin Exp Immunol. 2009;155:367-377.
4. Hsu D, Shaker M. An update on hereditary angioedema. Curr Opin Pediatr. 2012;24:638-646.
5. Kanani A, Schellenberg R, Warrington R. Urticaria and angioedema. Allergy Asthma Clin Immunol. 2011;7(suppl 1):S9.
6. Bowen T, Cicardi M, Farkas H, et al. 2010 international consensus
algorithm for the diagnosis, therapy and management of hereditary
angioedema. Allergy Asthma Clin Immunol. 2010;6:24.
7. Bernstein JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32:408-412.
8. Georgy MS, Pongracic JA. Hereditary and acquired angioedema. Allergy Asthma Proc. 2012;33(suppl 1):S73-S76.
9. Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474-481.
10. Cinryze (C1 esterase inhibitor [human]) product information. Exton, PA: ViroPharma Biologics, Inc; March 2013.
11. Berinert (C1 esterase inhibitor [human]) product information. Kankakee, IL: CSL Behring LLC; July 2012.
12. Kalbitor (ecallantide) product information.
Burlington, MA: Dyax Corp; February 2012.
13. Firazyr (icatibant) product information. Lexington, MA: Shire Orphan Therapies, Inc; August 2011.
14. Lexicomp Online. Danazol. Lexi-Drugs.
www.crlonline.com/lco/action/doc/retrieve/docid/patch_f/6690#pha.
Accessed February 28, 2013.
15. Lexicomp Online. Tranexamic acid. Lexi-Drugs.
www.crlonline.com/lco/action/doc/retrieve/docid/patch_f/7798. Accessed
February 28, 2013.
16. Norman JL, Holmes WL, Bell WA, Finks SW. Life-threatening ACE inhibitor-induced angioedema after 11 years on lisinopril. J Pharm Pract. 2012[epub]; doi:10.1177/0897190012465990.
To comment on this article, contact rdavidson@uspharmacist.com.


