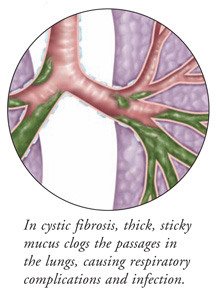
 Cystic fibrosis is an inherited disease in which a defective gene causes bodily fluids such as mucus, saliva, digestive juices, and sweat to become thick and sticky. When mucus is not thin and watery, it plugs the airways in the lungs, making it difficult to cough out. One of the first symptoms of cystic fibrosis is often chronic lung infection, since bacteria can easily grow in this thick mucus. The mucus also clogs the ducts of the pancreas, stopping the pancreatic enzymes that break down food from entering the intestines. Without these enzymes, many nutrients cannot be absorbed properly, and patients have a good appetite and bulky stools, but lose weight. Another problem is an increase in the amount of salt in the sweat, which can cause an imbalance of electrolytes in the blood while in a hot environment.
Cystic fibrosis is an inherited disease in which a defective gene causes bodily fluids such as mucus, saliva, digestive juices, and sweat to become thick and sticky. When mucus is not thin and watery, it plugs the airways in the lungs, making it difficult to cough out. One of the first symptoms of cystic fibrosis is often chronic lung infection, since bacteria can easily grow in this thick mucus. The mucus also clogs the ducts of the pancreas, stopping the pancreatic enzymes that break down food from entering the intestines. Without these enzymes, many nutrients cannot be absorbed properly, and patients have a good appetite and bulky stools, but lose weight. Another problem is an increase in the amount of salt in the sweat, which can cause an imbalance of electrolytes in the blood while in a hot environment.
All patients with cystic fibrosis do not have the same degree of physical problems. Some with mild disease are not diagnosed until their teenage years, while others with a more serious form are diagnosed a few days after birth.
Currently, there is no cure for this genetic defect, but modern treatments have significantly prolonged patients’ lifespans. The most common cause of death is respiratory failure, a result of thick mucus that blocks airways and causes chronic lung infections and scarred lung tissue.
Managing Complications of Cystic Fibrosis
In the United States, about 30,000 people suffer from cystic fibrosis, with 1,000 new cases diagnosed each year. Although years ago most children diagnosed with the disease did not reach elementary school age, today patients are likely to live into their late 30s and beyond.
Causes, Symptoms, and Diagnosis:
Cystic fibrosis is an inherited disease caused by a recessive gene. There are 10 million carriers of the cystic fibrosis gene in the U.S., and many are not aware they carry it because they have no symptoms of the disease. This gene changes an important protein that controls the movement of salt and fluid in and out of cells, causing an imbalance of salt in the body. This results in thick mucus that clogs the passages in the lungs and pancreas, and in salty sweat that can cause problems in hot environments.
Frequent cough, respiratory infections, shortness of breath, wheezing, and sinus infections are the primary symptoms of cystic fibrosis. As respiratory complications are the most common cause of death in these patients, aggressive treatment to keep the lungs clear of mucus and free of infection is critical.
The mucus also stops the digestive enzymes from moving out of the ducts of the pancreas. These enzymes are needed to break down fat and protein in the intestines, so without them, nutrients and many vitamins in food are not absorbed. Often patients have diarrhea or greasy stools. Cystic fibrosis causes patients to become thin and malnourished even though they have good appetites. Enzyme supplements must be taken with every meal to help digestion and maintain nutrient absorption. An inflammation of the pancreas, called pancreatitis, can result when the pancreatic ducts are blocked by mucus. With an increase in the amount of salt lost in the sweat, a hot environment can easily cause dehydration.
Diagnosis is done with a simple test of the amount of salt in the patient’s sweat, called the sweat test. Blood tests to identify the defective gene that causes cystic fibrosis may also be ordered. Since patients have many chronic, low-grade respiratory and sinus infections, x-rays of the lungs and sinuses are used to determine if inflammation or scarring is present. A culture of the sputum can show if there are bacteria present in the lungs and helps determine which antibiotics would be best to treat an infection.
Treatment Options:
The general goal of treatment is to minimize the complications of cystic fibrosis, since there is no cure at present. In order to keep the lungs healthy, infection-free, and clear of mucus, patients use daily chest physical therapy, exercise, and antibiotics. Chest physical therapy consists of pounding or clapping on the chest (percussion) to loosen the mucus, usually when the patient is sitting or lying in a position that will help the mucus drain (postural drainage). There are mechanical percussion machines that help loosen mucus, inflatable vests that help force the mucus out of the lungs, and breathing devices that cause vibrations to loosen the mucus. Exercise is also very helpful.
Oral and inhaled antibiotics are used to control the chronic bacteria present in the mucus of the lungs, and IV antibiotics are used when serious respiratory infections occur. Other inhaled medications, such as mucolytics and hypertonic saline, can be used to thin out mucus and make it easier to cough out. High doses of ibuprofen can be used to lessen inflammation in the lungs. Although ibuprofen is available OTC, it should be prescribed since the drug has several side effects and blood levels must be monitored by a doctor.
Digestive problems in cystic fibrosis are treated with the use of pancreatic enzyme supplements, taken before every snack and meal. These enzyme supplements help the body absorb the vitamins and nutrients in food and prevent malnutrition. Supplements of the fat-soluble vitamins (A, D, E, and K) are also needed to ensure enough are absorbed in the intestines. Some patients may have a feeding tube placed in their stomach overnight in order to receive liquid nutritional supplements.
Patients with cystic fibrosis should be treated by specialists in the disease. Your pharmacist can answer any questions you may have about the medications used to control the symptoms of cystic fibrosis.



