US Pharm.
2008;33(1)(Oncology suppl):21-28.
ABSTRACT:
Tumor lysis syndrome (TLS), an oncologic emergency, results when cancer cells
are rapidly lysed after chemotherapy or other treatments. Patients at risk
should be rapidly identified and therapeutic measures initiated. Goals are to
prevent or treat electrolyte anomalies and to preserve organ function.
Effective preventive strategies can minimize TLS and complications even in
highest-risk patients.
Tumor lysis syndrome (TLS) is
an oncologic emergency frequently encountered in clinical practice. It is
characterized by a spectrum of metabolic derangements often including
hyperuricemia, hyperphosphatemia with associated hypocalcemia, and
hyperkalemia, which occur as a result of rapid cellular lysis of cancer cells.
This syndrome is potentially fatal if left untreated; if inappropriately
managed, it can impart numerous medical complications and impair organs and
systems such as the kidneys, heart, central nervous system, and
musculoskeletal system.1
The management of patients at
risk for TLS begins with identifying those patients most likely to develop the
complication. Initiation of preventive strategies to avoid the metabolic
abnormalities and acute renal failure associated with TLS is essential for
positive patient outcomes. Health care practitioners responsible for the care
of cancer patients should have a strong understanding of TLS and how to manage
patients throughout the high-risk period. This review will provide an overview
of the pathophysiology of TLS, current therapeutic strategies to decrease the
likelihood of its development, and some medical interventions aimed at
ameliorating complications associated with TLS.
Epidemiology
TLS was first
reported almost 80 years ago, but its incidence remains ill-defined.1
The most commonly referenced percentages are from Hande and Garrow's 1993
retrospective analysis of 102 adult patients with high-grade non-Hodgkin's
lymphoma.2 Hande and Garrow reported the incidence of
TLS--identified through serial measurements of laboratory values--to be 42%,
whereas the incidence of clinically significant TLS was only 6% in the same
population. A similar occurrence rate has been demonstrated in pediatric
patients. Wossman et al reported the incidence of TLS to be 26.4% in children
with B-cell acute lymphoblastic leukemia.3 Reasons for the
inability to precisely define TLS incidence include variations in defining the
syndrome, variations in anticipating and studying its development in select
patient populations, and failure to report all occurrences.4
Despite the failure to
accurately pinpoint TLS prevalence in cancer patients, recognizing risk
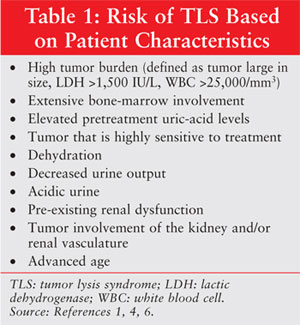
factors for its development is vital for managing patients. Predisposing
factors can be divided into patient characteristics (table 1), tumor types
(table 2), and treatment modalities.1,4-6 High-risk populations
include patients with a high tumor burden and an underlying diagnosis of a
highly proliferative tumor, particularly when highly sensitive to definitive
treatment consisting of chemotherapy, radiotherapy, hormonal therapy, and/or
biological therapy.4-6 TLS sometimes occurs spontaneously prior to
treatment initiation and in patients without recognizable risk factors.
7,8
Syndrome Summary
Pathophysiology1,4-6,9,10:
The hallmark pathogenesis of TLS is rapid cell lysis following the
administration of cytotoxic therapies. The large amount of intracellular
components dumped into the extracellular compartment exceeds the catabolic and
excretory capacities of the liver and kidneys. This sharp increase in the
concentration of selected cellular components overwhelms the body's normal
homeostatic mechanisms, resulting in impaired organ function (such as renal
failure) and associated morbidity (such as cardiac dysrhythmias and tetany).
The characteristic metabolic anomalies of TLS include hyperuricemia,
hyperphosphatemia with associated hypocalcemia, and hyperkalemia.
Definition:
Although there is a broad, universally accepted definition of TLS, a uniform,
discrete description of this syndrome is lacking. Two schemes to accurately
define TLS have been published that characterize TLS according to laboratory
features as well as clinical manifestations. The first formal definition was
published in 1993 by Hande and Garrow.2 Although it is the most
widely referenced definition of TLS, it has some limitations. First, the
definition requires that patients exhibit a 25% change in baseline laboratory
values, which does not account for those patients with abnormal values at the
time of presentation. Second, changes must occur within 96 hours after the
initiation of definitive treatment. Patients who develop TLS prior to
treatment or beyond four days of therapy are not covered by this definition.
A modified version of Hande
and Garrow's definition was published by Cairo and Bishop in 2004.4
The goal was to provide clinicians with a clinically relevant definition of
TLS that is practical and reproducible, like Hande and Garrow's version.
However, Cairo and Bishop broadened the time frame for TLS development
(abnormal values and changes could occur from three days before initiating
therapy to up to seven days after starting therapy) in an effort to capture
more cases; they also stratified patients according to low versus high risk.
Clinical Manifestations
and Consequences:
Clinical signs and symptoms associated with TLS may include nausea, vomiting,
lethargy, edema, congestive heart failure, dysrhythmias, muscle cramps,
tetany, paresthesias, back pain, syncope, renal failure, and seizures.
Although symptoms may develop upon patient presentation, they are more likely
to manifest within 12 to 72 hours after administration of cancer therapy.
1,4,6 Identification of patients experiencing TLS based solely on
observation of these signs and symptoms is not advisable since these
manifestations may be attributable to the patient's underlying malignancy.
Hyperuricemia, which typically
occurs two to three days after initiation of cytotoxic therapy, is a result of
the rapid release and catabolism of intracellular nucleic acids.1,4-6,11
Uric acid is the end product of purineñnucleic-acid catabolism by the enzyme
xanthine oxidase. Under normal physiologic conditions, more than 99% of uric
acid in the blood is in the soluble ionized form and uric acid is excreted by
the kidneys at a rate of 500 mg/day.4 Secretion of uric acid occurs
distal to the renal proximal tubule, as the solubility and excretion of uric
acid is favored in alkaline environments. Due to higher-than-normal
concentrations of nucleic acids in cancer cells compared with normal cells,
the destruction of malignant cells can overwhelm the kidneys' excretory
capacity of uric acid. If uric acid remains at high concentrations in acidic
conditions, uric-acid crystals may form in the distal tubules and the
collecting ducts, resulting in obstructive uropathy and uremia. Conditions
that can compound the development of uric-acid crystallization include
dehydration, decreased glomerular filtration rate, and acidosis.1,4,6,9

Hyperphosphatemia is seen when
intracellular phosphorus of malignant cells, which can be as much as four
times the concentration in normal cells, is released to the extracellular
compartment.4 Although the kidneys initially respond to the
increase in phosphorus concentrations with increased urinary excretion and
decreased tubular reabsorption, in time the tubular transport mechanisms
become saturated and hyperphosphatemia occurs, usually 24 to 48 hours
following initial cellular destruction. The most significant complication
resulting from increased concentrations of phosphorus is the formation of
calcium phosphate precipitates in the renal tubules, resulting in acute renal
failure. Hyperphosphatemia is frequently found in association with
hypocalcemia since calcium and phosphorus homeostasis are closely and
reciprocally linked.1,4,9
The most serious consequence
of TLS is hyperkalemia, which usually is seen within six to 72 hours after
cytotoxic therapy is initiated. Liberation of intracellular potassium into the
extracellular space can quickly overwhelm the kidneys' ability to excrete
potassium. High serum concentrations of potassium, which can be exacerbated by
renal failure, acidosis, and hypocalcemia, can lead to ventricular arrhythmias
and sudden death. Symptomatic patients should be evaluated for dialysis, as
this is the most effective method of lowering serum potassium values.
1,4,9
The development of acute renal
failure in the setting of TLS is frequently the result of uric-acid
nephropathy and volume depletion. As previously mentioned, patients with TLS
can experience precipitation of uric-acid crystals in the kidneys. Volume
depletion in the cancer patient is seen frequently and is often
multifactorial. Volume depletion can be related to disease or diagnostic
evaluation, and reasons for it include vomiting, diarrhea, poor oral intake,
insensible losses, and fasting prior to procedures and tests. When a decrease
in intravascular volume occurs, the patient may experience a prerenal state,
which in turn can lead to increases in tubular uric-acid concentrations.
1,4,9
Prevention and Treatment
The most effective
management strategies target TLS prevention and sequelae associated with TLS.
Health care providers must be proactive and a bit overcautious in identifying
at-risk patients and initiating protective measures. Attempts to correct
metabolic disturbances, especially those that are life-threatening or could
prevent initiation of cytotoxic therapy, should be made in a timely fashion.
Aggressive
Hydration/Forced Diuresis/Discontinuation of Medications1,4,9,10:
The first measure undertaken to prevent TLS involves the provision of
adequate hydration to 1) increase intravascular volume, thereby decreasing
extracellular concentrations of uric acid, potassium, and phosphorus; and 2)
enhance renal blood flow to maintain aÜ sufficient glomerular filtration rate
and urine output. Ideally, aggressive hydration should be started at least 24
to 48 hours prior to chemotherapy initiation; however, this may not always be
feasible, depending on tumor type or the patient's clinical condition. IV
hydration should be instituted at a rate to provide patients with 2 to 3 L/m
2/day, which is approximately twice maintenance fluids. The goal of
increased hydration, which should continue through the duration of initial
cancer therapy, is to maintain urine output at a rate of greater than 100
mL/hour (2-3 mL/kg/hour for pediatric patients) and urine specific gravity of
less than 1.010. In patients at high risk for fluid overload (e.g., the
elderly, congestive heart failure, renal failure, and so on), fluids should be
started, but at a more conservative rate.
If adequate urine output
cannot be achieved with IV hydration alone, diuretics may be used to achieve
the desired outcome. They also can be used to prevent fluid overload in
patients and assist with potassium excretion. Loop diuretics, such as
furosemide, are used most commonly to achieve these effects. An alternative
diuretic is mannitol (0.5 g/kg/dose IV q 6-8 h).
Another measure that health
care providers must institute is the discontinuation of agents that may worsen
the patient's condition should he or she experience TLS. Electrolyte
supplementation, particularly potassium and phosphorus products, should be
discontinued and removed from IV fluids. Medications known to cause
electrolyte disturbances--such as angiotensin-converting enzyme inhibitors,
which can increase potassium values--should be discontinued and alternative
therapeutic recommendations provided to the health care team. Additionally,
medications proven to be nephrotoxic (e.g., aminoglycosides, NSAIDs, and the
like) should be avoided, if possible, during high-risk periods.
Urinary Alkalinization
1,4,9,10:
Alkalinization of the urine assists in decreasing the incidence of uric-acid
nephropathy and subsequent renal failure by reducing uric-acid
crystallization. In alkaline environments (pH >7.0), uric acid remains ionized
(in the form of urate); thus, it is more water-soluble and more readily
excreted by the kidneys. Methods of alkalinizing the urine include the
addition of sodium bicarbonate or sodium acetate to IV fluids or the
administration of oral acetazolamide (Diamox); both methods require monitoring
of patients for signs and symptoms of metabolic alkalosis.
Selection of which method to
use to increase urinary pH remains controversial. In general, urinary
alkalinization can induce the formation of calciumñphosphate precipitates in
the renal microvasculature and tubules. This can cause an obstructive
nephropathy that can ultimately increase the chance of acute renal failure by
decreasing the glomerular filtration rate. Urinary alkalization can cause a
xanthine nephropathy by decreasing the solubility of xanthine, a precursor of
uric acid. The use of sodium bicarbonate systemically can lower the amount of
circulating calcium by strengthening calciumñphosphate bonding, further
exacerbating the hypocalcemia seen in TLS.
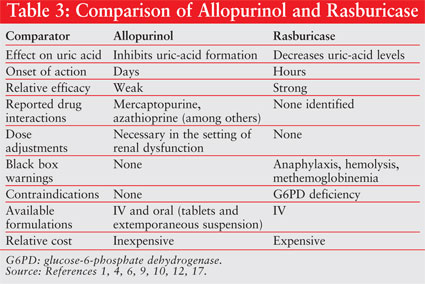
Allopurinol
1,4,9,10,11: A
structural analog of hypoxanthine, allopurinol is a competitive inhibitor of
xanthine oxidase, an enzyme necessary for purine catabolism. When converted to
its active metabolite, oxypurinol, allopurinol prevents the conversion of
hypoxanthine to xanthine and xanthine to uric acid. Although it is effective
in preventing the formation of new uric acid and the incidence of uric-acid
obstructive uropathy, it has no effect on existing uric-acid levels. Thus, it
usually takes at least two to three days from the initiation of allopurinol
before a reduction in uric acid is achieved.
Due to its inhibition of
xanthine oxidase, allopurinol has some clinical limitations. First, its use
may result in a xanthine nephropathy. When the enzyme responsible for the
catabolism of xanthine and its precursors is blocked, an increase in the
concentration of these compounds is often problematic. Second, allopurinol can
interact with medications that rely on xanthine oxidase for their metabolism
(e.g., mercaptopurine, azathioprine). Dose reductions of such medications must
be made if they are to be used concomitantly with allopurinol.
Urate Oxidase:
The enzyme urate oxidase is endogenous to mammals except humans and most
primates. Also known as uricase, it is the enzyme responsible for the
catabolism of uric acid to allantoin, which is five to 10 times more soluble
in urine than uric acid. Uricozyme, a nonrecombinant form of urate oxidase
isolated from Aspergillus flavus, has been used in Europe since the
mid-1970s. Uricozyme has proven efficacy in lowering uric-acid levels more
rapidly than in historic controls who had received allopurinol in combination
with hyperhydration. However, its use is limited by potential immunogenicity
and declining efficacy through production of antiuricase antibodies as well as
by serious side effects including allergic reactions, anaphylaxis, and
bronchospasm.4-6, 10,12
In 2002, the FDA approved a
recombinant form of urate oxidase, rasburicase (Elitek), isolated as a cDNA
clone from A flavus and expressed in Saccharomyces cerevisiae to
produce large quantities of purified protein.4-6,12,13 Rasburicase
has been proven safe and efficacious in several clinical trials.14-16
It rapidly lowers uric-acid levels, usually within four hours of
administration. compared with allopurinol, its ability to reduce uric-acid
levels is not only faster, but more substantial as well.15 For a
comparison of allopurinol and rasburicase, see table 3. Rasburicase is
generally well tolerated, the most frequently reported side effects being
nausea, vomiting, headache, and fever. It is, however, contraindicated in
patients with glucose-6-phosphate dehydrogenase deficiency due to reports of
hemolytic anemia and methemoglobinemia in these patients following its use.
4,6,12
One other disadvantage of
rasburicase is its high cost. It is approved for use at a dose of 0.15-0.2
mg/kg IV daily for five days.12 Some investigators have considered
alternative dosing regimens (such as fixed doses of rasburicase or shorter
duration of therapy) in an effort to address the high cost of the product.
6,17,18 These dosing schemes are not formally endorsed by the company's
product labeling, but they warrant further consideration.
Electrolyte Disturbances:
Electrolyte derangements
associated with TLS should be addressed in relation to the patient's clinical
status and handled on a case-by-case basis. Some electrolyte disturbances may
warrant monitoring only, whereas some aberrations may require expeditious
interventions to prevent serious morbidity and/or mortality. Each disturbance
has its own set of interventions aimed at normalization of serum values and
amelioration of clinical sequelae.
Summary
TLS can be fatal if untreated or
poorly managed, but clinicians have several effective therapeutic options for
managing cancer patients to prevent its occurrence. Patients at greatest risk
for TLS should be managed the most aggressively in an effort to achieve
positive outcomes. Associated electrolyte disturbances, although not
completely preventable, can be lessened if patients are monitored judiciously.
If TLS is treated appropriately, the risk of secondary complications, such as
renal failure, can be significantly reduced.
REFERENCES
1. Davidson M, Thakkar S, Hix JK, et al. Pathophysiology, clinical consequences, and treatment of tumor lysis syndrome. Am J Med. 2004;116:546-554.
2. Hande KR, Garrow GC. Acute tumor lysis in patients with high-grade non-Hodgkin's lymphoma. Am J Med. 1993;94:133-139.
3. W^ssman W, Schrappe M, Meyer U, et al. Incidence of tumor lysis syndrome in children with advanced stage Burkitt's lymphoma/leukemia before and after introduction of prophylactic use of urate oxidase. Ann Hematol. 2003;82:160-165.
4. Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
5. Jeha S. Tumor lysis syndrome. Semin Hematol. 2001;38(suppl 10):4-8.
6. Bessmertny O, Robitaille LM, Cairo MS. Rasburicase: a new approach for preventing and/or treating tumor lysis syndrome. Curr Pharm Des. 2005;11:4177-4185.
7. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors--a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
8. Gemici C. Tumour lysis syndrome in solid tumours. Clin Oncol (R Coll Radiol). 2006;18:773-780.
9. Rampello E, Fricia T, Malaguarnera M. The management of tumor lysis syndrome. Nat Clin Pract Oncol. 2006;3:438-447.
10. Coiffier B, Riouffol C. Management of tumor lysis syndrome in adults. Expert Rev Anticancer Ther . 2007;7:233-239.
11. Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87-114.
12. McEvoy GE, ed. AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2007.
13. Elitek [package insert]. New York, NY: Sanofi-Synthelabo Inc; January 2007.
14. Pui CH, Mahmoud HH, Wiley JM, et al. Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol. 2001;19:697-704.
15. Goldman SC, Holcenberg JS, Finklestein JZ, et al. A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis. Blood. 2001;97:2998-3003.
16. Jeha S, Kantarjian H, Irwin D, et al. Efficacy and safety of rasburicase, a recombinant urate oxidase (Elitek), in the management of malignancy-associated hyperuricemia in pediatric and adult patients: final results of a multicenter compassionate use trial. Leukemia. 2005;19:34-38.
17. Sood AR, Burry LD, Cheng DK. Clarifying the role of rasburicase in tumor lysis syndrome. Pharmacotherapy . 2007;27:111-121.
18. Trifilio S, Gordon L, Singhal S,
et al. Reduced-dose rasburicase (recombinant xanthine oxidase) in adult cancer
patients with hyperuricemia. Bone Marrow Transplant. 2006;37:997-1001.
To comment on this article, contact
editor@uspharmacist.com.


