US Pharm. 2008;33(7)(Oncology
suppl):14-22.
ABSTRACT:
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder
characterized by a translocation between chromosomes 9 and 22, forming the
Philadelphia (Ph) chromosome. This and other chromosomal abnormalities can be
detected with the use of cytogenetics, a branch of genetics focusing on
chromosomal structure. Standard cytogenetics, fluorescence in situ
hybridization (FISH), and reverse-transcriptase polymerase chain reaction
(RT-PCR) are cytogenetic methods employed to diagnose CML and to monitor
response to treatment. A pivotal trial demonstrating the superiority of
imatinib in patients with newly diagnosed CML reported complete cytogenetic
response, with no Ph-positive (Ph+) cells seen in a sample of bone marrow, as
a primary endpoint.
Chronic myelogenous leukemia
(CML) is a myeloproliferative disorder initiated by a genetic translocation
within a pluripotent stem cell. This malignant transformation leads to
unregulated growth and accumulation of myeloid cells.1,2 The
disease has a triphasic course, with 80% diagnosed in the chronic phase
(lasting an average of four to six years untreated), followed by the accelerated
phase, and finally, the blast crisis.3 Most patients are
asymptomatic when diagnosed. As the disease progresses, presentation may
include the clinical picture of a myeloproliferative disorder: leukocytosis,
thrombocytosis, anemia, weight loss, and splenomegaly.
New cases of CML are forecast
to total 4,830 in the United States in 2008, with 450 deaths.4 The
median age of onset is 67 years, although the disease occurs in all age groups.5
New treatment modalities such as tyrosine kinase inhibitors (i.e., imatinib,
dasatinib, nilotinib) are prolonging survival, but the only current therapy
that is potentially curative is allogeneic stem cell transplant. This is
problematic given the morbidity associated with transplant and the older
average age of patients with CML.5
Classification of CML
A number of
classifications are in use for the determination of the disease stage.
Increases in the number of blasts in the peripheral blood or bone marrow
(5%-19%, depending on the classification system) are a key event declaring
progression to the accelerated phase.5 Additional features may
include anemia or thrombocytopenia unrelated to therapy, rapid leukocyte
doubling time (<5 days), persistent thrombocytosis, splenomegaly, clonal
evolution, persistent fever, or bone pain.5 An increase in blast
count to above 20% to 30% marks progression to the final phase of CML--blast
crisis. This final phase is rapidly fatal, with a median survival of three to
six months if untreated.6 Treatment of CML is more successful in
chronic rather than the later accelerated or blast phases.7
In 1960, Nowell and Hungerford
developed a technique to disrupt cell mitosis and expand the cells using
hypotonic solution.8 During normal cell replication, the
chromosomes are tightly wound and impossible to visualize.9
Subsequent studies of the now more visible contents of the cell nuclei
uncovered a chromosomal abnormality (termed the Philadelphia [Ph] chromosome)
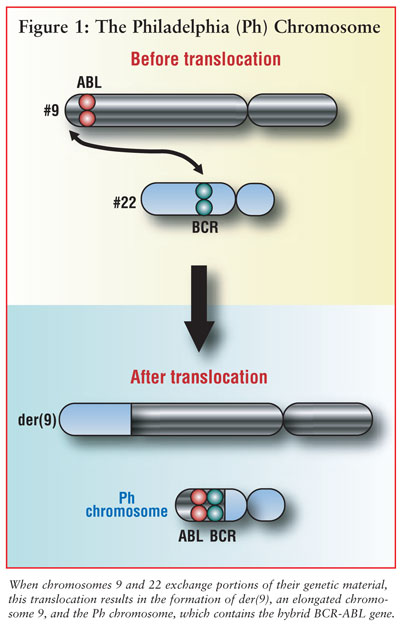
in the cells of seven patients with CML.10 This reciprocal exchange
of material between the Abelson kinase domain (ABL) of chromosome 9 and the
breakpoint cluster region (BCR) of chromosome 22 is seen in over 90% of
patients diagnosed with CML. The full nomenclature t(9;22)(q34;q11) designates
a translocation (t) between the two chromosomes at the short arm (q) of 9
(region 3, band 4) and the short arm of 22 (region 1, band 1).11
The hybrid BCR-ABL tyrosine kinase is constitutively active, dysregulating
downstream pathways driving malignant cell proliferation and resistance to
apoptosis.12 The pathway initiates with adenosine triphosphate
(ATP) binding to BCR-ABL. The formation of the Ph chromosome is shown in Figure
1. Imatinib, a small molecule tyrosine kinase inhibitor approved for the
first-line treatment of chronic phase CML in 2002, inhibits BCR-ABL signaling
by binding to the active form of ABL at the ATP-binding site.8
Cytogenetics and CML
Methods to reveal
chromosomal abnormalities enable the diagnosis and appropriate treatment of
CML.13 Cytogenetics, a branch of genetics focusing on chromosomal
structure, is used to determine the presence of genetic changes within
cellular DNA. Standard cytogenetics is usually performed on a bone marrow
sample due to the greater number of proliferating cells in bone marrow
compared to peripheral blood.12 Cell division is induced and then
chemically arrested in metaphase. Chromosomes are stained to increase
visibility (Giemsa or G-banding), demonstrating alternating dark and light
bands when viewed under the microscope.13
Conventional cytogenetics, or
karyotyping, is employed at presentation to identify the characteristic Ph
chromosome. The analysis is time consuming, typically involving the
examination of 25 to 30 metaphase cells.12 This method is
considered insufficiently sensitive to detect the presence of Ph-positive
(Ph+) chromosomes in less than 10% of cells. The same methodology can also be
used to monitor response to treatment at three-month intervals, defined by the
percent of dividing cells in the sample that retain the Ph marker.14,15
FISH and RT-PCR Testing:
Patients have an average of 1 x 1012 leukemic cells at presentation.13,16
After treatment, the lack of detection of Ph+ chromosomes using conventional
cytogenetic analysis is considered a complete cytogenetic response (CCR).
Patients in CCR have been shown to have up to 1 x 109 leukemic
cells still present.
To detect further log
reductions in disease burden requires tests of greater sensitivity.
Fluorescence in situ hybridization (FISH) involves the incubation of
fluorescence labeled DNA probes with denatured (single-strand) DNA.
Chromosomes have a distinct pattern of light absorption, related to the
density of the guanine-rich DNA. Traditional FISH, or dual-FISH analysis, uses
one colored probe fluorescing from the BCR region and a second different
colored probe fluorescing from the ABL region. In CML, the two regions are no
longer on different chromosomes but are superimposed, fluorescing the blended
color. These traditional FISH analyses have a false positive rate of up to
10%. Newer methods report false positives below 1%.12 In
hypermetaphase FISH, up to 500 cells are analyzed, yielding results with much
greater accuracy than conventional cytogenetics. This technique can be used to
analyze samples from blood, bone marrow, or tissue in any stage of cell
replication.13 FISH allows increased resolution of genetic
aberrations not detailed with standard cytogenetics and without the time
needed to culture cells.17 FISH analysis is used at some centers to
monitor maintenance of CCR.
Molecular studies with
reverse-transcriptase polymerase chain reaction (RT-PCR) begin with single
strands of DNA sequences specific to a disease. Subsequent cycles with a DNA
polymerase result in exponential multiplication of the target sequence.13
RT-PCR is the most sensitive of all monitoring tools with the ability to pick
up one CML cell in a population of 100,000 or more cells. In CML, quantitative
RT-PCR results report the ratio of the target BCR-ABL sequence to a reference
gene (BCR or ABL). RT-PCR results can be available faster than FISH, but have
lower specificity. False positive results can result from contamination of a
BCR-ABL negative sample by a positive sample. The rapid duplication of DNA
sequences (up to 30 cycles) will amplify any contamination that might be
introduced.12
FISH and RT-PCR are valuable
tools in the identification of individuals with Ph-negative BCR-ABL-positive
CML. Of the 5% to 10% of CML patients not demonstrating the Ph chromosome with
conventional cytogenetics, some may have submicroscopic BCR-ABL aberrations,
or more complex translocations in addition to the classic breakpoints of
chromosomes 9 and 22. Atypical Ph translocations are termed simple
(chromosome 22 plus a chromosome other than 9) or complex (9, 22, and
other chromosomes).18 Thirty to fifty percent of these cases of
"masked" CML are readily identifiable with the more sensitive molecular
methods.13 The remaining half of those Ph negative by cytogenetics
and molecular methods are termed atypical CML and have a poor prognosis.6
Molecular methods are also necessary for detection of low levels of Ph+ cells,
minimal residual disease, or 1 x 105 leukemic cells.16
Chromosomal Changes and
Mutations: Conventional
cytogenetic analysis is the only technique that allows visualization of all
chromosomes. This analysis is used for monitoring patients for acquisition of
additional chromosomal aberrations, which signal transition from chronic to
accelerated phase.14 Blood or marrow samples from patients
converting to accelerated or blast phase often demonstrate chromosomal
abnormalities in addition to the Ph chromosome, termed clonal evolution.
BCR-ABL down regulates the DNA repair process, enabling additional mutations.8
The most frequently seen chromosomal changes include trisomy 8, isochrome 17,
duplicate Ph chromosome, loss of 17p and BCR-ABL1. Other changes seen less
frequently include trisomy 19, trisomy 21, trisomy 17, and deletion 7.19
For patients in CCR, RT-PCR monitoring is recommended at three-month
intervals, with conventional cytogenetics repeated annually.15
Mutations in the BCR-ABL gene
can occur as well. Determination of the specific mutation is clinically
important as it may dictate a change in therapy. Changes in the structure of
BCR-ABL can interfere with drugs designed to inhibit downstream signaling.
Over 40 different mutations involving amino acid substitutions have been
reported, most often found in the area where ATP binds to BCR-ABL. This area
has been termed the P loop. G250E, Q252H, Y253F, and E255K mutations in
the P loop are not involved in the binding of imatinib, but have been linked
with poor prognosis and a survival of four to five months.19 The
"gatekeeper" mutation T315I interferes with imatinib binding, predicting
resistance to imatinib and other tyrosine kinase inhibitors (i.e., dasatinib
and nilotinib). Sequencing of the BCR-ABL gene, specifically the ABL kinase
domain, to look for mutations may be performed if there is suboptimal response
to initial therapy, loss of response to therapy, or progression to a more
advanced stage of CML.
Targeted Therapies:
Tyrosine Kinase Inhibitors
Currently, response
to therapy in CML is defined on three levels. Hematologic response
describes counts of cells in peripheral blood samples. Complete
hematological response (CHR) equates to normalization of the blood counts. Cytogenetic
response is determined by the percent of Ph+ chromosomes remaining in the
bone marrow (complete = no Ph+ cells; partial = up to 34% Ph+ cells; minor =
35%-90% Ph+ cells).5 In addition, a major molecular response
is reflective of a three-log
reduction in the number of CML-defining, BCR-ABL genes.20,21
As previously described,
formation of the tyrosine kinase Ph chromosome in CML results in unchecked
growth of malignant cells. Tyrosine kinase inhibitors are a class of agents
that interfere with the downstream signaling initiated by these abnormal
proteins. These molecules block the transfer of phosphate from adenosine
triphosphate, interfering with tyrosine kinase mediated signaling pathways and
the subsequent proliferation of leukemic cells. The three tyrosine kinase
inhibitors currently available for treatment in the U.S. are imatinib,
dasatinib, and nilotinib (Table 1).
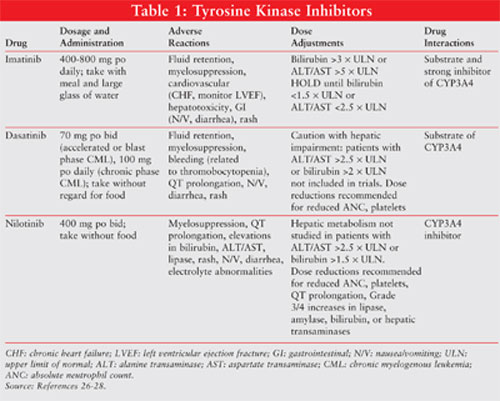
Imatinib:
In the pivotal trial comparing the efficacy of imatinib, an oral tyrosine
kinase inhibitor, to interferon alfa plus low-dose cytarabine in patients with
newly diagnosed CML, cytogenetic response was found to be associated with
event-free survival. The International Randomized Study of Interferon and
ST1571 (IRIS) trial reported that of patients demonstrating a CCR at 12
months, only 3% had progressed to accelerated or blast phase in 60 months. In
the group not achieving a major cytogenic response in 12 months, 19%
progressed to accelerated or blast phase by 60 months.22 Based on
these outcome data and availability of sensitive methods used to monitor
response to treatment, clinicians are able to change therapy if a response is
not evident by 12 months following the initiation of treatment.
Many of the definitions and
time frames for response arise from experience with imatinib. Patients with suboptimal
response might demonstrate normal blood counts, but have no cytogenic
response at three months, greater than 35% Ph+ cells at six months, greater
than 5% of Ph+ cells at 12 months, or no molecular response following 18
months of treatment. Guidelines recommend for patients exhibiting suboptimal
response with standard treatment doses of imatinib (400 mg daily) that if
tolerated, the dose may be escalated to 600 to 800 mg daily.23
Resistance
to imatinib is defined as no hematologic response after three months of
treatment, no cytogenetic response after six months of treatment, greater than
35% Ph + after 12 months, or greater than 5% Ph+ cells after 18 months of
treatment. Those fitting the definition of resistance are considered to have
failed imatinib, and the treatment should be changed.23 In the IRIS
study, 4% of newly diagnosed CML patients became resistant yearly, with the
number reducing to fewer than 2% in year 5 of the study.22 However,
it is estimated that 40% of those in late chronic phase and 70% to 90% in
accelerated or blast phase are resistant to imatinib.21
The IRIS study also
demonstrated the prognostic value of cytogenetic changes in CML. All patients
achieving a major molecular response by 12 months of imatinib treatment had
progression-free survival at 60 months.21,22
Dasatinib:
One treatment consideration for patients failing imatinib is the multitargeted
tyrosine kinase inhibitor, dasatinib. Dasatinib inhibits BCR-ABL in both the
active and inactive formation, as well as other downstream proteins, including
c-KIT, platelet-derived growth factor receptor, and the Src family of kinases.
The diversity of binding mechanisms available to dasatinib contributes to its
ability to inhibit the constitutive activity of the Ph chromosome in patients
with mutations that prevent activity of imatinib.
The Phase II START (SRC/ABL
Tyrosine kinase inhibitor Activity Research Trials of dasatinib) trials tested
the efficacy of dasatinib in different CML populations.24 In
START-C, patients with CML in chronic phase with resistance to imatinib were
treated with 70 mg of dasatinib twice daily. Most patients (90%) achieved a
CHR. In the START-A arm, 107 accelerated-phase CML patients with primary or
acquired resistance to imatinib (n = 99), or intolerance to imatinib therapy
(n = 8), received 70 mg dasatinib twice daily. At the onset of this study, 60%
of participants exhibited BCR-ABL mutations. At eight months follow-up, 87% (n
= 81) achieved an overall hematologic response (major and minor hematologic
responses), and 24% (n = 26) achieved a CCR. START-R was a randomized trial
comparing dasatinib 70 mg twice daily to imatinib 800 mg daily in
chronic-phase CML patients resistant to imatinib. A major cytogenetic response
was attained in 32% of dasatinib patients and 4% of imatinib patients after 12
weeks of therapy. Of note, dasatinib was not effective in patients with the
T315I mutation.24
Nilotinib:
Recently approved in October 2007, nilotinib is another tyrosine kinase
inhibitor effective against many imatinib-resistant mutations. In a phase II
trial, 119 patients with imatinib resistant or intolerant accelerated-phase
CML received 400 mg of nilotinib twice daily. Hematologic responses were
achieved in 47% of patients. Major cytogenetic response was achieved in 29% of
patients. Like dasatinib, nilotinib was active in most patients with mutations
conferring resistance to imatinib, with the exception of the T315I mutation.25
Potential Therapies
Therapies under
development to treat patients resistant to tyrosine kinase inhibitors include
aurora kinase inhibitors, which are active against BCR-ABL demonstrating the
T315I mutation. Farnesyl transferase inhibitors and P13-K inhibitors target
the BCR-ABL pathway downstream of the ATP binding site and are unaffected by
T315I mutations.21
Conclusion
The presence of a
specific genetic abnormality, targeted drug therapies, and the availability of
sensitive and specific methods to monitor response make CML a unique disease
amongst hematologic malignancies. Recent advances allow clinicians to make
adjustments to therapy often before overt changes in the clinical course
occur. Notably, knowledge of the molecular pathways and events driving disease
progression are leading to the development of novel drug therapies.
REFERENCES
1. Hill R. The
biology of cancer. In: Rubin P, ed. Clinical Oncology. 8th ed.
Philadelphia, PA: WB Saunders Company; 2001:32-45.
2. McGuire T, Pavletic
S. Chronic leukemias. In: Dipiro J, Talbert R, Yee G, et al, eds. Pharmacotherapy:
A Pathophysiologic Approach. 5th ed. New York, NY: McGraw Hill;
1999:2397-2408.
3. Drucker B, Lee S.
Chronic leukemias. In: DeVita V, Hellman S, Rosenberg S, eds. Cancer
Principles & Practice of Oncology. 7th ed. Philadelphia, PA:
Lippincott Williams & Wilkins; 2005:2121-2133.
4. Jemal A, Siegel R,
Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96.
5. National
Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology
V3.2008. Chronic myelogenous leukemia. www.nccn.org. Accessed March 19, 2008.
6. Ghelani D, Sneed T,
Bueso-Ramos C, et al. Chronic myeloid leukemia. In: Kantarjian H, Wolff R,
Koller C, eds. MD Anderson Manual of Medical Oncology. New York, NY:
McGraw-Hill; 2006.
7. Radich J, Dai H, Mao
M, et al. Gene expression changes associated with progression and response in
chronic myelogenous leukemia. Proc Natl Acad Sci USA.
2006;103:2794-2799.
8. Wong S, Witte O. The
BCR-ABL story: bench to bedside and back. Annu Rev Immunol.
2004;22:247-306.
9. Moore C, Best R.
Chromosome preparation and banding. Encyclopedia of Life Sciences.
April 19, 2001.
http://mrw.interscience.wiley.com/emrw/9780470015902/els/article/a0001444/current/html.
Accessed March 14, 2008.
10. Nowell P. Discovery
of the Philadelphia chromosome: a personal perspective. J Clin Invest.
2007;117:2033-2035.
11. Mitelman F,
Johansson B, Mertens F. The impact of translocations and gene fusions on
cancer causation. Nat Rev Cancer. 2007;7:233-245.
12. Wang Y, Bagg A,
Pear W, et al. Chronic myelogenous leukemia: laboratory diagnosis and
monitoring. Genes Chromosomes Cancer. 2001;32:97-111.
13. Tefferi A, Dewald
G, Litzow M, et al. Chronic myeloid leukemia: current application of
cytogenetics and molecular testing for diagnosis and treatment. Mayo Clin
Proc. 2005;80:390-402.
14. Kantarjian H,
Schiffer C, Jones D. Monitoring the response and course of chronic myeloid
leukemia in the modern era of BCR-ABL tyrosine kinase inhibitors: practical
advice on the use and interpretation of monitoring methods. Blood.
2008;111:1774-1780.
15. Hughes T, Deininger
M, Hochhaus A, et al. Monitoring CML patients responding to treatment with
tyrosine kinase inhibitors: review and recommendations for harmonizing current
methodology for detecting BCR-ABL transcripts and kinase domain mutations and
for expressing results. Blood. 2006;108:28-37.
16. Sessions J. Chronic
myeloid leukemia in 2007. J Manag Care Pharm. 2007;13(suppl A):4-7.
17. Ried T.
Cytogenetics--color and digitized. N Engl J Med.
2004;350:1597-1600.
18. Babicka L, Zemanova
Z, Pavlistova L, et al. Complex chromosomal rearrangements in patients with
chronic myeloid leukemia. Cancer Genet Cytogenet. 2006;168:22-29.
19. Quintas-Cardama A,
Cortes J. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc.
2006;81:973-988.20. Sawyers CL. Chronic myeloid leukemia. N Engl J Med.
1999;340:1330-1340.
21. Kantarjian H, Giles
F, Quintas-Cardama A, et al. Important therapeutic targets in chronic
myelogenous leukemia. Clin Cancer Res. 2007;13:1089-1097.
22. Druker B, Guilhot
F, O'Brien S, et al. Five-year follow-up of patients receiving imatinib for
chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
23. Baccarani M, Saglio
G, Goldman J, et al. Evolving concepts in the management of chronic myeloid
leukemia: recommendations from an expert panel on behalf of the European
Leukemianet. Blood. 2006;108:1809-1820.
24. Talpaz M, Shah N,
Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia
chromosome-positive leukemias. N Engl J Med. 2006;354:2531-2541.
25. le Coutre P,
Ottmann O, Giles F, et al. Nilotinib (formerly AMN107), a highly selective
BCR-ABL tyrosine kinase inhibitor, is active in patients with
imatinib-resistant or intolerant accelerated-phase chronic myelogenous
leukemia. Blood. 2008;111:1834-1839.
26. Gleevec (imatinib)
package insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation;
September 2007.
27. Sprycel (dasatinib)
package insert. Princeton, NJ: Bristol-Myers Squibb Company; November 2007.
28. Tasigna (nilotinib)
package insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation;
October 2007.
To comment on this article, contact rdavidson@jobson.com.



