US Pharm. 2011;36(1)(Oncology suppl):3-6.
ABSTRACT: Cancer is a worldwide health problem. In the United States, it is the second leading cause of death. Cancer is a heterogeneous disease that is characterized by the loss of cellular controls through the accumulation of genetic and epigenetic changes. Genetic changes are caused by alterations in DNA, including mutations that contribute to aberrant cellular functions. Epigenetic changes, however, are hereditable changes in gene expression without a change in the DNA sequence. Currently, four epigenetic agents (vorinostat, romidepsin, azacitidine, decitabine) have been approved by the FDA for the treatment of hematologic malignancies, specifically cutaneous T-cell lymphoma (CTCL) and myelodysplastic syndromes (MDS). Hematologic malignancies are types of cancers that affect blood, bone marrow, and lymph nodes. These cancers account for about 9% of new cancer diagnoses in the U.S. This article provides a conceptual overview of the use of epigenetic agents in the intervention and management of hematologic malignancies.
Cancer remains the second leading cause of death in the United States. It is estimated that approximately 1.5 million new cases of cancer were diagnosed in 2009, costing the U.S. government more than $220 billion.1 Cancer is a heterogeneous disease that is caused by aberrant genetic and epigenetic changes that allow the cell to escape normal controls. Traditionally, it was viewed that cancer is a result of genetic alterations. Genetic alteration can damage the DNA structure and induce mutations. Conventional chemotherapeutic agents interfere with cellular functions, including inhibition of DNA synthesis and blockage of cell division. However, these agents are toxic and/or have minimum therapeutic value, necessitating exploration of novel anticancer agents that exert their effects through an alternative mode of action.2,3
Epigenetic changes affect gene expression without directly changing DNA sequences. In eukaryotic cells, approximately 2 meters of DNA are compacted inside the 6-µm–diameter nucleus. This DNA compaction is facilitated by small, basic histone proteins. Histones interact with negatively charged phosphate groups of DNA through their positively charged side chains arginine and lysine. About 146 base pairs of DNA are wrapped around an octamer, composed of two copies of each core histone protein (histones H2A, H2B, H3, and H4). This octameric nucleosome unit occurs in about every 200 base pairs of eukaryotic genomes.4 Nucleosomes are the basic structural and functional unit of chromatin, which is the complex of DNA and proteins in eukaryotic cells.4 Linker DNA joins the nucleosomes together, which gives the appearance of beads on a string when viewed under electron microscope.4
Each core histone protein has a structured and an unstructured amino-terminal tail domain. The tail domain protrudes from the nucleosome core. Histone tails provide sites for posttranslational modifications, including acetylation, methylation, and phosphorylation.5
Among all of the posttranslational modifications on amino-terminal histone tail domains, histone acetylation has been extensively studied. Acetylation is a reaction catalyzed by histone acetyltransferases (HATs), and the reverse deacetylation reaction is catalyzed by histone deacetylases (HDACs). These families of enzymes regulate the states of chromatin, which correlate with gene expression. The addition of an acetyl group removes the positive charge on lysine, thus neutralizing the basic charge of the histone. This modification is suggested to reduce the affinity between histones and DNA, which in turn correlates with gene expression. Acetylated histone is usually associated with transcriptionally active chromatin.6 Deacetylation of histones, however, is associated with transcriptional repression.6 Genes are turned “on” or “off” via posttranslational modifications on histone tails of chromatin.6 Consistent with this view, HDACs are overexpressed in cancers of varied tissue origin, such as breast and prostate.6-9 Histone deacetylase inhibitors (HDACIs) target cancer cells overexpressing deacetylases and have little effect on normal cells.8,9 Currently, two HDACIs, vorinostat (Zolinza) and romidepsin (Istodax), have been approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL).
In addition to posttranslational modifications on histone proteins, DNA methylation plays a major role in the initiation and maintenance of cancer. DNA methylation is catalyzed by a group of enzymes called DNA methyltransferases (DNMTs). It occurs in DNA at CpG dinucleotides.7 More than 60% of genes are associated with CpG dinucleotides in their promoter regions.7 CpG dinucleotides are generally unmethylated, which correlates with active gene expression.7,10 On the other hand, CpG dinucleotides are underrepresented in the genome. In cancer cells, DNA methylation in the genome is loss (hypomethylation) and CpG dinucleotides in the promoter regions are methylated (hypermethylation).7,10 Currently, two DNA hypomethylating (demethylating) agents, namely azacitidine (Vidaza) and decitabine (Dacogen), have been approved by the FDA for the treatment of myelodysplastic syndromes (MDS).
HEMATOLOGIC MALIGNANCIES
Hematologic malignancies are types of cancers that affect blood, bone marrow, and lymph nodes. According to the Leukemia & Lymphoma Society, it is estimated that 137,260 people in the U.S. will be diagnosed with leukemia, lymphoma, or myeloma in 2010.11 New cases of these malignancies will account for approximately 9% of all new cancer cases diagnosed in the U.S. this year.11
Cutaneous T-Cell Lymphoma
CTCL is a malignancy of T lymphocytes primarily manifesting in the skin.12 It is a rare and heterogeneous type of cancer. CTCL accounts for about 3% of all non-Hodgkin’s lymphomas (NHLs) and affects mostly adults.13 CTCL cells express many of the same surface molecules as normal counterparts, such as CD45R0. Mycosis fungoides (indolent lymphoma) is the most common type of CTCL. It appears as skin patches and usually progresses over many years. Sézary syndrome is an advanced (aggressive) form of mycosis fungoides.12,14
Clinical prognostic indicators of CTCL include the level of skin surface involvement, the level of lymph node involvement, dissemination to visceral sites, and the presence of CTCL cells in the circulation.12 These variables are used in the modified TNM (tumor, node, metastasis) staging classification, as shown in TABLE 1.
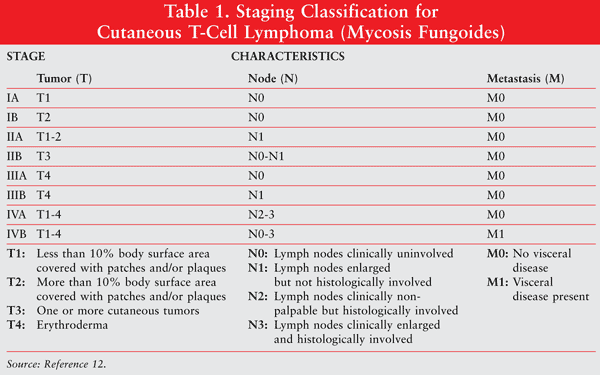
Principles of Therapy: Treatment varies according to the type and stage of the disease. Common treatment modalities include skin-directed therapy (e.g., radiotherapy, phototherapy, topical chemotherapy, and total skin electron-beam therapy), systemic therapy, and combined strategies. Early-stage disease has an excellent chance of cure or long-term control.12 Topical therapy with corticosteroids is the preferred modality for early-stage disease.12 Late-stage disease is rarely cured but can be palliated. Systemic therapy is preferred for patients with extensive disease.14
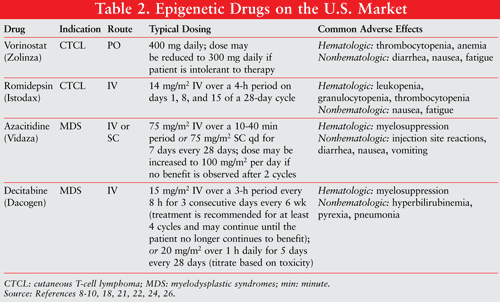
Although these modalities are effective, most patients rapidly become refractory to conventional agents, thus necessitating exploration of novel anticancer agents that exert their effects through an alternative mode of action. Although the precise mechanism is still unknown, interest in epigenetic therapy is expanding and this modality holds great promise as a treatment approach against cancer. Among the various agents, HDACIs represent a new class of molecules for CTCL. Deacetylation of histones is associated with transcriptional repression.6,7 As of 2010, two HDACIs—vorinostat (Zolinza) and romidepsin (Istodax)—have been approved by the FDA for CTCL (TABLE 2).
Vorinostat (Zolinza) is an FDA-approved HDACI for the treatment of CTCL in patients with progressive, persistent, or recurrent disease on or after treatment with two systemic therapies. Vorinostat is a hydroxamic acid that targets HDAC class I and class IIa/IIb.8,9 Hematologic malignancies are associated with hypoacetylation, which correlates with inactivation of genes responsible for apoptosis, cell-cycle arrest, and tumor suppression. This epigenetic agent is relatively selective toward cancer cells.8-9
The major trial that provided evidence for the FDA’s approval of the drug was a single-arm, open-label, multisite, nonrandomized study evaluating the efficacy of vorinostat in 74 patients with stage IB and higher CTCL who had failed two systemic therapies, one of which was bexarotene.15-18 Each patient received a 400-mg oral dose of vorinostat 7 days per week. The dose could be reduced for toxicity to 300-mg oral dose for 5 days per week. Criteria for evaluating response to vorinostat treatment were changes from baseline in the proportion of skin patches, plaques, or tumors to the body surface area, lymph node size, extent of blood involvement, and pruritus.15-18 The primary efficacy end point was objective disease response. The overall objective response rate (complete response and partial response) was 30% in all patients treated with vorinostat, and one patient with stage IIB had a complete clinical response. With the exception of one patient, all responses were partial.17,18 The estimated median response duration was 168 days.15-18 The most common nonhematologic adverse effects were fatigue (52%), diarrhea (52%), and nausea (41%). Serious adverse effects were thrombocytopenia and severe anemia.15-18
Overall, vorinostat is efficacious for patients with CTCL. It is also well tolerated. This HDACI is currently being investigated for its efficacy and safety in various types of cancers and in other treatment modalities, including combination therapy. The approved dosage is 400 mg orally once daily. Vorinostat is supplied as 100-mg capsules.19
Romidepsin (Istodax) is an FDA-approved HDACI for the treatment of CTCL in patients who have received at least one prior systemic therapy. It was approved in 2009. Romidepsin is a cyclic peptide that targets HDAC class I.8,9 The major trials that provided pivotal evidence for the FDA’s approval were two single-arm, open-label, multisite studies.20,21 The efficacy of romidepsin was evaluated in 167 patients. Criteria for evaluating response to romidepsin were composite end points that included assessments of skin, lymph node, and visceral involvement, and presence of Sézary cells. The primary efficacy end point for both trials was the overall response rate. The overall response rate was 34% in study 1 and 35% in study 2. The complete response was 6% in both trials. The median response duration was 15 months in study 1 and 11 months in study 2. Common adverse effects in trials were nausea and fatigue. The serious adverse effects were leukopenia, granulocytopenia, and thrombocytopenia. The recommended dose for romidepsin is 14 mg/m2 intravenously over 4 hours on days 1, 8, and 15 of a 28-day cycle (TABLE 2).20,21
Myelodysplastic Syndromes
MDS are a group of bone marrow disorders in which the bone marrow does not produce enough healthy blood cells. MDS affect mostly the elderly, with the median age of onset between 60 and 70 years of age.12,22 These diseases are more common in males. The exact etiology of MDS is unknown. These syndromes are characterized by cytopenia and bone marrow dysplasia.12,22 General symptoms of MDS are low red cell count (anemia), low white cell count (neutropenia), and low platelet count (thrombocytopenia).22
Several classification systems have been developed for MDS. Historically, the French-American-British (FAB) classification system has been used. In this classification system, patients with MDS are categorized into five subtypes based on the percentage of blood and marrow blasts, the presence of marrow sideroblasts, the number of peripheral monocytes, and the presence of Auer rods.12,23 The newer classification system suggested by the World Health Organization expands the categories of MDS subtypes.23
The International Prognostic Scoring System (IPSS) is used for grading the severity of patients with MDS. This scoring system is based on the percentage of marrow blasts, the presence of cytogenetic abnormalities, and the degree of cytopenia. In the IPSS system, patients are divided into low, intermediate-1, intermediate-2, and high-risk groups. For patients with intermediate-2 or high risk, the median overall survival is 1.2 years or 0.4 years, respectively.23 The IPSS system has been incorporated into clinical trial design for MDS.
Principles of Therapy: There are several general approaches to treatment, including transfusions, iron chelation therapy, chemotherapy, administration of growth factors, and drug therapy. At present, three drugs have been approved by the FDA for the treatment of MDS: the immunomodulatory agent lenalidomide and the DNA hypomethylating agents azacitidine and decitabine (TABLE 2). Azacitidine and decitabine are predominately used for higher-risk MDS patients.10
Azacitidine is indicated for use in patients with all five FAB subtypes of MDS. The safety and efficacy of azacitidine were shown in a phase III, multicenter, randomized trial in 191 patients.24 Patients were randomized to receive 75 mg/m2 azacitidine subcutaneously daily for 7 days every 28 days or best supportive care. Patients who received best supportive care were permitted to cross over to azacitidine treatment. The primary efficacy end point was the response rate. The overall response rate was 23% (7% complete response, 16% partial response) in all patients treated with azacitidine.24 Compared with patients receiving best supportive care, MDS patients had durable hematologic improvements, including increases in red blood cells, white blood cells, platelet numbers, and transfusion independence. Among the 99 patients in the azacitidine treatment arm, 51% had a red blood cell lineage response.24 The most common hematologic adverse effect was myelosuppression, which decreased with the onset of a response. The most common nonhematologic side effects were injection site reactions, diarrhea, nausea, and vomiting (TABLE 2).22,24,25
Decitabine, another DNA hypomethylating agent, is indicated for the treatment of all MDS subtypes and intermediate-1, intermediate-2, and high-risk IPSS groups. It is given intravenously. The pivotal clinical trial that led to the approval of this drug showed that the overall response rate was 17% (9% complete response, 8% partial response) in the 89 decitabine-treated patients versus 0% for 81 patients receiving standard supportive care.26 The adverse effects of decitabine were well tolerated (TABLE 2).26-28
CONCLUSION
Hematologic malignancies account for about 9% of new cancer diagnoses in the U.S. However, conventional cancer modalities are either ineffective or toxic to normal cells. As a result, epigenetic agents were investigated for the treatment of these disorders. Epigenetic drugs work by inhibiting a cellular process that silences the genes involved in controlling the development of cancer. This means that epigenetic agents can reverse certain genes, which were repressed in cancer cells, that play important roles in cancer suppression. At present, the FDA has approved four epigenetic drugs for the treatment of hematologic malignancies. Even though these epigenetic agents have made a significant improvement in the therapeutic management of hematologic malignancies, complete remission rates and overall response rates remain relatively low. Thus, the exploration of novel epigenetic anticancer agents, combinations of the approved agents, and targeted drug-delivery systems to enhance safety and efficacy are essential to improve patient outcomes.
REFERENCES
1. American Cancer Society. Cancer Facts & Figures 2009. Atlanta, GA: American Cancer Society; 2009.
2. Hanahan D, Bergers G, Bergsland E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045-1047.
3. Morgan G, Ward R, Barton M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin Oncol. 2004;16:549-560.
4. Luger K, Mader AW, Richmond RK, et al.
Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251-260.
5. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074-1080.
6. Buchwald M, Krämer O, Heinzel T. HDACi—targets beyond chromatin. Cancer Lett. 2009;280:160-167.
7. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity. 2010;105:4-13.
8. Hoshino I, Matsubara H. Recent advances in histone deacetylase targeted cancer therapy. Surg Today. 2010;40:809-815.
9. Marks PA. Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim Biophys Acta. 2010;1799:717-725.
10. Mani S, Herceg Z. DNA demethylating agents and epigenetic therapy of cancer. Adv Genet.
2010;70:327-340.
11. The Leukemia & Lymphoma Society. Facts 2010-2011. White Plains; NY: Leukemia & Lymphoma Society; 2010.
12. DeVita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
13. Lansigan F, Choi J, Foss FM. Cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:979-996.
14. Gardner JM, Evans KG, Musiek A, et al. Update on treatment of cutaneous T-cell lymphoma. Curr Opin Oncol. 2009;21:131-137.
15. Duvic M, Olsen EA, Breneman D, et al. Evaluation of the long-term tolerability and clinical benefit of vorinostat in patients with advanced cutaneous T-cell lymphoma. Clin Lymphoma Myeloma. 2009;9:412-416.
16. Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31-39.
17. Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109-3115.
18. Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247-1252.
19. Zolinza (vorinostat) package insert. Whitehouse Station, NJ: Merck & Co, Inc; February 2010.
20. Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410-5417.
21. Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485-4491.
22. List AF. Treatment strategies to optimize clinical benefit in the patient with myelodysplastic syndromes. Cancer Control. 2008;15(suppl):2-3.
23. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis of myelodysplastic syndromes. Blood. 1997;89:2079-2088.
24. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429-2440.
25. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223-232.
26. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794-1803.
27. Steensma DP, Baer MR, Slack JL, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27:3842-3848.
28. Wijermans PW, Lubbert M, Verhoef G, et al. An epigenetic approach to the treatment of advanced MDS; the experience with the DNA demethylating agent 5-aza-2’-deoxycytidine (decitabine) in 177 patients. Ann Hematol. 2005;84(suppl 1):9-17.
To comment on this article, contact rdavidson@uspharmacist.com.



