US Pharm. 2014;39(5):HS2-HS8.
ABSTRACT: Pediatric patients with nephrotic syndrome (NS) can exhibit proteinuria, hypoalbuminemia, edema, and dyslipidemia. Morbidity and mortality associated with nephrotic syndrome, including progression to end-stage renal disease (ESRD), are reduced with treatment. Idiopathic NS occurs more often than NS from secondary causes in pediatric patients. Corticosteroids are used first-line to induce remission and are effective in most pediatric patients. Inadequate response to corticosteroids dictates the need for adjunctive or alternative therapies such as alkylating agents, calcineurin inhibitors, mycophenolate mofetil, and rituximab. These strategies are useful for patients who do not remit with steroids, who frequently relapse, or who are corticosteroid-dependent.
Pediatric patients with nephrotic syndrome (NS) have increased glomerular filtration barrier permeability, resulting in clinical features such as proteinuria, hypoalbuminemia, edema, and dyslipidemia.1,2 The annual incidence and prevalence of NS in children are two to seven cases per 100,000 and 12 to 16 cases per 100,000, respectively.2 In children, idiopathic NS (INS) occurs more often than NS from to secondary causes such as diabetes and systemic lupus erythematosus.3 With biopsy, INS can be diagnosed by histopathologic type to predict treatment success and prognosis, although the treatment strategy is unlikely to differ.
ETIOLOGY, DIAGNOSIS, AND OUTCOMES
The most prevalent pathology of INS in children is minimal-change disease (MCD). A much smaller, but rising, percentage of INS is attributed to focal segmental glomerulosclerosis (FSGS). Diffuse mesangial hypercellularity, membranous nephropathy, membranoproliferative glomerulonephritis, and immunoglobulin A (IgA) nephropathy are less common.4 Symptom onset in patients with MCD commonly occurs from 2 to 6 years of age.2 Corticosteroid responsiveness is the most significant prognostic factor in INS.2 Children with steroid-sensitive nephrotic syndrome (SSNS) are more likely to have favorable long-term outcomes, whereas those with steroid-resistant nephrotic syndrome (SRNS) have comparatively lower renal survival rates.1 Approximately 90% of pediatric patients with INS achieve complete remission (CR) (TABLE 1) with corticosteroids and are classified as having SSNS (TABLE 2).2 Therefore, renal biopsy is not required for all children, owing to the high likelihood of response to initial and subsequent courses of corticosteroids.1
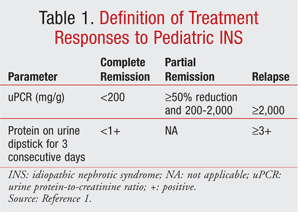

Renal biopsy is indicated in children with SSNS who fail to respond to corticosteroids after one or more remissions or who have deteriorating renal function while receiving calcineurin inhibitors (CNIs), despite dose reduction.1 High suspicion of other etiologies, such as FSGS based on older age or the higher prevalence in African or African American individuals, may warrant biopsy. Pediatric patients with SRNS (TABLE 2) should undergo biopsy to exclude secondary causes of NS and to evaluate severity. Secondary causes also can be excluded through medical history, physical examination, laboratory tests, and imaging. Although genetic mutations have been identified in some children with SRNS and FSGS, routine evaluation is not recommended because of the high cost and lack of utility with regard to prognosis and treatment modification at this point.
In children, proteinuria is defined as urine protein excretion ≥40 mg/m2/h, a protein-creatinine ratio (PCR) ≥2,000 mg/g, an albumin-creatinine ratio ≥300 mg/dL, or 3+ on urine dipstick.1 In the absence of overt clinical symptoms, nephrotic-range proteinuria is generally defined as urine PCR ≥2,000 mg/g. Quantification of proteinuria is used as a surrogate marker of outcomes and is important for assessing prognosis and treatment response.
COMPLICATIONS AND ASSOCIATED THERAPIES
In addition to proteinuria, other complications of glomerular disease, such as hypertension, edema, thrombosis, and infection, should be treated to reduce associated morbidity and mortality. Adequate dietary protein intake (0.8-1.0 g/kg/day) should be maintained and is optimally utilized in conjunction with a high-carbohydrate diet.1 For children with IgA nephropathy, an ACE inhibitor (ACEI) or angiotensin receptor blocker (ARB) is suggested for proteinuria between 0.5 and 1 g/day per 1.73 m2.1 For other pediatric INS etiologies, renin-angiotensin system blockade is not recommended for proteinuria independent of hypertension. However, an ACEI or ARB is recommended first-line for children with hypertension regardless of the degree of proteinuria.5 The target blood pressure for children with glomerulonephritis is <50th percentile for both systolic and diastolic pressure for age and sex.5
Although it is debated whether a reduction in hyperlipidemia positively impacts renal function, children with NS should typically be classified and treated as high-risk patients owing to the association with cardiovascular (CV) disease.1 Childhood lipid levels are representative of future lipid levels and CV events in a general pediatric population.6 In children with chronic kidney disease, it is recommended that lipid levels be evaluated annually owing to the influence of growth and development. Statins are typically not recommended for dyslipidemia in children with NS, owing to inadequate assessment of clinically relevant outcomes and long-term safety data in this population. Emphasis is placed on therapeutic lifestyle changes, including dietary fat restriction and weight loss, when appropriate.
Additional dietary modifications include moderate sodium restriction in patients with edema associated with NS.1 Diuretic therapy may also be required, although even patients with a normal glomerular filtration rate may be resistant to diuretics. Loop diuretics are used most commonly and may be given in combination with thiazide diuretics or metolazone to augment effects. Intravenous (IV) diuretic therapy may be needed in patients who are unresponsive to oral therapy from possible effects of intestinal-wall edema on gastrointestinal absorption. Pediatric patients receiving diuretic therapy should be monitored cautiously because of the increased risk of hypovolemia in this population.
Thrombophilic risks should be considered in children with NS.1 Although venous thromboembolism (VTE) occurs in only 3% of pediatric NS cases, it may be associated with significantly increased morbidity and mortality.7 Serum albumin <2.5 g/dL has been associated with an increased risk of thrombosis in adults, although this association was not found in a large cohort of pediatric patients.1,7 Risk factors for VTE development in children include worsening proteinuria and histologic evidence of membranous nephropathy and/or class V lupus nephritis. No definitive recommendations have been made concerning the pharmacologic prevention of VTE in pediatric patients with NS. Nonpharmacologic risk-reduction strategies should be implemented, and pharmacologic prophylaxis may be considered in high-risk groups.7
Children with NS should be immunized for prevention of infection, including the pneumococcal influenza vaccination.1,8 Live vaccines are contraindicated while patients are receiving immunosuppressants and should be deferred until the prednisone dose is <1 mg/kg daily (<20 mg/day) or 2 mg/kg on alternate days (<40 mg on alternate days).1,8
TREATMENT OF INS
Corticosteroids
Although the exact mechanism is unknown, corticosteroid therapy has been highly effective for pediatric NS for nearly 5 decades, with as few as 20% of patients failing to respond.1,9,10 Greater success with corticosteroids occurs in NS that is due to MCD than in NS that is due to FSGS.10
Randomized, controlled trials (RCTs) have not established corticosteroid dosing; however, the International Study of Kidney Disease in Children recommends oral prednisone 60 mg/m2/day or 2 mg/kg/day (maximum dose 60 mg/day) once daily for 4 to 6 weeks, followed by 40 mg/m2 or 1.5 mg/kg (maximum 40 mg on alternate days) every other day for 2 to 5 months, with tapering.1,11,12 Alternatively, prednisolone may be given at the same doses.1 Clinical studies correlate longer and higher doses of corticosteroids with higher rates of remission and a lower incidence of relapse; thus, regimens with corticosteroid exposure less than what is presented should not be used.13 Nevertheless, extended-duration dosing (i.e., 6 months) results in relapse in nearly 40% of patients.13 Results from two currently registered placebo-controlled trials evaluating corticosteroid administration for durations of 20 weeks and 24 weeks are anticipated.
Children who relapse after initially responding to corticosteroids will likely respond to corticosteroids again and should be treated with a subsequent dose of oral prednisone 60 mg/m2 or 2 mg/kg (maximum 60 mg/day) until CR has been maintained for 3 days.1 Thereafter, alternate-day dosing of corticosteroids should be continued for 4 weeks. Although adverse effects (AEs) in clinical trials did not increase with longer durations (i.e., 5-7 months) of corticosteroid therapy compared with a shorter duration (i.e., 2 months), repetitive treatments could reasonably be expected to increase the risk of weight gain, growth impediments, hypertension, hyperglycemia, bone loss, psychological disorders, and adrenal suppression.13 Children who do not remit after corticosteroid therapy are classified as SRNS and are at greater risk for mortality and ESRD.10 Low-quality evidence supports the use of high-dose corticosteroids in SRNS.1 Additionally, children with SSNS can be deemed late nonresponders, usually within the first year, if they exhibit resistance or partial response to a minimum of 4 weeks of full-dose corticosteroids; these patients may derive benefit from nonsteroidal therapies.14
Alternative Agents
Nonsteroidal agents may be used in pediatric patients to reduce exposure to corticosteroids, thereby decreasing related AEs, or when steroid resistance is demonstrated. These medications, which are referred to as corticosteroid-sparing agents when used for frequent-relapsing NS (FRNS) or steroid-dependent NS (SDNS), include alkylating agents, CNIs, mycophenolate mofetil (MMF), and rituximab. Predictors of FRNS include initial remission time ≥9 days and first relapse in ≤6 months.15 Recommendations related to the use of these agents for FRNS and SDNS, as well as data for SRNS, are presented below.
Alkylating Agents (Cyclophosphamide, Chlorambucil): Alkylating agents can reduce relapse in pediatric NS by 24% to 72% up to 5 years after remission, consequently lowering corticosteroid exposure in patients with SSNS classified as FRNS or SDNS.1,16 Although these agents’ immunosuppressant effects are well known, the exact mechanism in NS has not been defined. The use of oral cyclophosphamide (2 mg/kg/day) for 8 to 12 weeks (maximum cumulative dose 168 mg/kg) or oral chlorambucil (0.1-0.2 mg/kg/day) for 8 weeks (maximum cumulative dose 11.2 mg/kg) in children is supported by moderate-quality evidence.1 Although hemorrhagic cystitis is unlikely to occur when recommended doses are used, patients should be in remission prior to cyclophosphamide initiation in order to tolerate large volumes of fluid and produce adequate urine output to protect against the risk of this disorder.1 Subsequent exposure to alkylating agents is not recommended, owing to the risk of dose-dependent gonadal toxicity (in both males and females). Reduced sperm counts have been reported with cumulative doses of cyclophosphamide >200 to 300 mg/kg and chlorambucil 8 to 10 mg/kg.1 Cyclophosphamide, which is metabolized through glutathione S-transferase, may be more efficacious in patients expressing this polymorphism.17 Chlorambucil may be associated with a higher risk of cancer, seizures, and bacterial infections versus cyclophosphamide.1,18 Current recommendations include no role for alkylating agents in SRNS, based on limited evidence suggesting that the risks far outweigh the benefits.1
CNIs (Cyclosporine, Tacrolimus): The recommended doses of CNIs for pediatric NS are oral cyclosporine 4 to 5 mg/kg/day (starting dose) divided twice daily and oral tacrolimus 0.1 mg/kg/day (starting dose) divided twice daily for 12 months.1 Given their relatively similar efficacy, there is no order of preference in using alklyating agents versus CNIs for FRNS or SDNS.1 However, unlike alkylating agents, CNIs may be used as initial therapy for SRNS. If the patient fails to achieve partial remission or CR at 6 months, discontinuation of the CNI may be considered; otherwise, the CNI should be continued for at least 12 months.1 Most studies have examined CNIs given in addition to low-dose corticosteroids for SRNS; however, superiority has not been established between CNI monotherapy and CNI in combination with corticosteroids.1 AEs associated with CNIs include hypertension, renal impairment, and hyperkalemia.1,10 Diabetes mellitus also has been associated with tacrolimus use in children.1 Monitoring for trough levels of 80 to 150 ng/mL for cyclosporine and 5 to 10 ng/mL for tacrolimus may be targeted to minimize nephrotoxicity.1 Although most studies on SRNS have been performed using cyclosporine, tacrolimus is an alternative for patients unwilling to experience hypertrichosis and gum hypertrophy, which are associated with long-term (>1 year) cyclosporine use.10 CNIs are very expensive, and although not currently recommended, one study demonstrated that using 50% less cyclosporine provided similar efficacy and safety via coadministration of ketoconazole 1.5 mg/kg/day as a metabolic inhibitor of cyclosporine.19
MMF: Although MMF is probably less effective than CNIs, a few small pediatric studies show promising results with MMF in maintenance of remission from SDNS and FRNS, with minimal AEs.1 Oral MMF doses used in studies were 1,200 mg/m2/day or about 30 mg/kg/day in two divided doses.1 The high cost of MMF is likely to be a limitation, and therapy should not be discontinued sooner than 12 months to avoid the risk of relapse. In SRNS, MMF is an option for patients who fail to achieve remission with CNI and steroids.1,10
Rituximab: Of all the alternative agents, rituximab has the most limited evidence for use in SDNS, SRNS, and FRNS. Studies have used rituximab 375 mg/m2 IV once weekly for 1 to 4 weeks. Rituximab is believed to work by acting on the B cell, but it may have a more direct effect on the podocyte itself in FSGS.20 Current guidelines recommend rituximab only in children with SDNS who have continuing frequent relapses despite optimal combinations of prednisone and corticosteroid-sparing agents, and/or who have serious AEs from therapy.1 The high cost also must be considered. Based on the lack of RCTs and the concern over AEs, rituximab is not recommended for SRNS.1
Other Agents: Moderate-quality evidence supports the use of levamisole 2.5 mg/kg on alternate days for 1 year to reduce the risk of relapse in pediatric SSNS.1 However, the manufacturer voluntarily discontinued production of levamisole partly due to the risk of agranulocytosis.1,21 Consequently, outside of veterinary use, the drug is no longer available in the United States. Aside from corticosteroids, adrenocorticotropic hormone (H.P. Acthar Gel) is the only FDA-approved agent for the treatment of NS without uremia of the idiopathic type or due to lupus erythematosus.9,22
Galactose, adalimumab, and thiazolidinediones are potential future therapeutic options for pediatric NS.10 Until larger trials evaluate these agents’ utility, no dosing strategies or targeted populations can be recommended. Mizoribine and azathioprine are not recommended because RCTs show no additional benefit of either drug over placebo in FRNS or SDNS.1
CONCLUSION
Corticosteroids continue to be the mainstay of treatment for pediatric NS and have a high degree of success, although relapse often occurs. Steroid-sparing agents are available for pediatric patients experiencing frequent relapses or steroid dependence. Alkylating agents, CNI, MMF, and rituximab are beneficial in SSNS, whereas fewer options exist for SRNS. A limited number of RCTs demonstrate varying benefit with each of these agents, and an accompanying set of disadvantages such as short-term and long-term AEs, cost, and required monitoring for each drug class must be considered.
REFERENCES
1. Kidney Disease: Improving Global Outcomes (KDIGO)
Glomerulonephritis Work Group. KDIGO clinical practice guideline for
glomerulonephritis. Kidney Int Suppl. 2012;2:139-274.
2. Ulinski T, Aoun B. New treatment strategies in idiopathic nephrotic syndrome. Minerva Pediatr. 2012;64:135-143.
3. Elie V, Fakhoury M, Deschênes G, Jacqz-Aigrain E. Physiopathology
of idiopathic nephrotic syndrome: lessons from glucocorticoids and
epigenetic perspectives. Pediatr Nephrol. 2012;27:1249-1256.
4. Filler G, Young E, Geier P, et al. Is there really an increase in non-minimal change nephrotic syndrome in children? Am J Kidney Dis. 2003;42:1107-1113.
5. Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure
Work Group. KDIGO clinical practice guideline for the management of
blood pressure in chronic kidney disease. Kidney Int Suppl. 2012;2:337-414.
6. Kidney Disease: Improving Global Outcomes (KDIGO) Lipid Work
Group. KDIGO clinical practice guideline for lipid management in chronic
kidney disease. Kidney Int Suppl. 2013;3:259-305.
7. Kerlin BA, Haworth K, Smoyer WE. Venous thromboembolism in pediatric nephrotic syndrome. Pediatr Nephrol. June 28, 2013 [Epub ahead of print].
8. Beck L, Bomback AS, Choi MJ, et al. KDOQI US commentary on the
2012 KDIGO clinical practice guideline for glomerulonephritis. Am J Kidney Dis. 2013;62:403-441.
9. Koskimies O, Vilska J, Rapola J, Hallman N. Long-term outcome of primary nephrotic syndrome. Arch Dis Child. 1982;57:544-548.
10. Greenbaum LA, Benndorf R, Smoyer WE. Childhood nephrotic syndrome—current and future therapies. Nat Rev Nephrol. 2012;8:445-458.
11. Arneil GC. The nephrotic syndrome. Pediatr Clin North Am. 1971;18:547-559.
12. Alternate-day versus intermittent prednisone in frequently
relapsing nephrotic syndrome. A report of “Arbetsgemeinschaft für
Pädiatrische Nephrologie.” Lancet. 1979;1:401-403.
13. Hodson EM, Willis NS, Craig JC. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev. 2007;(4):CD001533.
14. Straatmann C, Ayoob R, Gbadegesin R, et al. Treatment outcome of
late steroid-resistant nephrotic syndrome: a study by the Midwest
Pediatric Nephrology Consortium. Pediatr Nephrol. 2013;28:1235-1241.
15. Nakanishi K, Iijima K, Ishikura K, et al; Japanese Study Group of
Renal Disease in Children. Two-year outcome of the ISKDC regimen and
frequent-relapsing risk in children with idiopathic nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8:756-762.
16. Gipson DS, Massengill SF, Yao L, et al. Management of childhood onset nephrotic syndrome. Pediatrics. 2009;124:747-757.
17. Vester U, Kranz B, Zimmermann S, et al. The response to
cyclophosphamide in steroid-sensitive nephrotic syndrome is influenced
by polymorphic expression of glutathion-S-transferases-M1 and -P1. Pediatr Nephrol. 2005;20:478-481.
18. Latta K, von Schnakenburg C, Ehrich JH. A meta-analysis of
cytotoxic treatment for frequently relapsing nephrotic syndrome in
children. Pediatr Nephrol. 2001;16:271-282.
19. Iyengar A, Kamath N, Phadke KD, Bitzan M. Cyclosporine/ketoconazole reduces treatment costs for nephrotic syndrome. Indian J Nephrol. 2013;23:419-423.
20. Fornoni A, Sageshima J, Wei C, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011;3:85ra46.
21. Wolford A, McDonald TS, Eng H, et al. Immune-mediated
agranulocytosis caused by the cocaine adulterant levamisole: a case for
reactive metabolite(s) involvement. Drug Metab Dispos. 2012;40:1067-1075.
22. H.P. Acthar Gel (repository corticotropin injection) product
information. Union City, CA: Questcor Pharmaceuticals, Inc; January
2006.
To comment on this article, contact rdavidson@uspharmacist.com.



