US Pharm. 2014;39(11)(Specialty&Oncology suppl):8-11.
ABSTRACT: Acute myeloid leukemia (AML), the most frequently occurring form of leukemia, is associated with a spectrum of mutations. One of these mutations, FMS-like tyrosine kinase 3 (FLT3), is a receptor tyrosine kinase that activates downstream pathways of cellular proliferation and antiapoptosis. Mutations often occur in the FLT3 enzyme, making it an appealing target for specific inhibitors. However, monotherapy with FLT3 inhibitors has not produced satisfactory clinical outcomes and has additionally led to drug resistance. Several novel FLT3 inhibitors are under investigation for their utility in AML. Based on current literature, possible strategies for treating AML could be either multikinase FLT3 inhibitors or an FLT3 inhibitor combined with traditional chemotherapy.
Acute myeloid leukemia (AML) is an aggressive malignancy characterized by the accumulation of abnormal precursor myeloid cells that interfere with the development and function of leukocytes, erythrocytes, and platelets.1-5 These cells can relocate to the peripheral blood circulation, as well as infiltrate the lungs and cerebrospinal fluid.3 The primary diagnostic feature of AML is a >20% myeloblast composition in the blood, with exceptions to the threshold.1,3
Mutations in AML
AML is an extremely heterogeneous disease associated with numerous genetic (molecular marker) and cytogenetic (chromosomal karyotype) abnormalities upon presentation.1,4 Cytogenetic abnormalities, such as translocation (t) and inversion (inv), may confer either favorable (e.g., t[15;17], inv[16], t[16;16], t[8;21]) or poor (e.g., inv[3], t[9;22], t[1;22], t[8;16]) outcomes.4 The presence of Auer rods (usually with t[8;21]) and surface proteins CD13/CD33 also suggest myeloid cell deformities.1,3,4
There are multiple activating mutations in AML (e.g., RAS, c-KIT, JAK2). The 2008 World Health Organization (WHO) classification emphasizes examining at least three important oncogenes: nucleophosmin-1 (NPM1), FMS-like tyrosine kinase 3 (FLT3), and CCAAT/enhancer-binding protein alpha (CEBPA).1,3,4,6 These molecular markers are now routinely tested in patients with newly diagnosed AML.1,3,4 As depicted in FIGURE 1, the normally occurring or wild-type FLT3 gene encodes for a class III receptor tyrosine kinase (RTK) that activates downstream enzymes (including, but not limited to, STAT5, AKT, MEK, and NF-kB) and eventually enhances cell proliferation and antiapoptosis.1,4,5,7,8
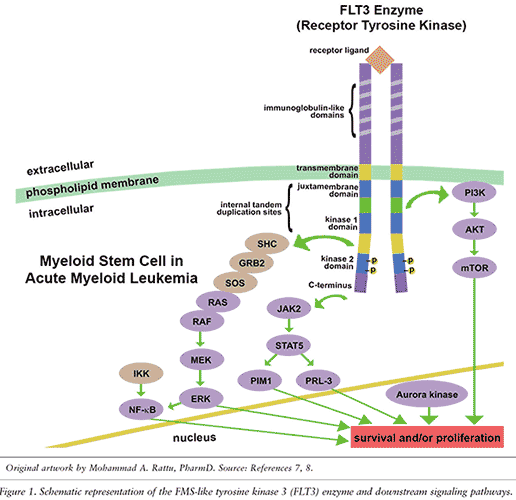
The FLT3 enzyme can be overexpressed and/or develop gain-of-function mutations in the tyrosine kinase domain (TKD), as well as an internal tandem duplication (ITD) of 3 to 400 base pairs in the juxtamembrane and/or TKD1 domains.1,4,7,8 While each of these mutations enhances the constitutive action of the FLT3 enzyme and promotes cellular proliferation, the FLT3-ITD mutation is associated with an inferior prognosis, especially when a patient is homozygous for the oncogene.1,4,5,7-9 NPM1 mutations are the most common and they confer relatively better outcomes with chemotherapy alone.1,4,5,9 Concomitant mutations in NPM1 and FLT3-ITD negate the improved prognosis of an NPM1 mutation.1,4
In addition, a monosomal karyotype (MK) denotes an incurable AML, regardless of an allogeneic hematopoietic cell transplantation (HCT).3 Patients with a normal karyotype (NK) and a NPM1 or CEBPA (but no FLT3-ITD) mutation have a prognosis (up to a 70% cure rate) similar to patients with the most favorable cytogenetics (i.e., inv[16] or t[8;21]).3 NK patients with FLT3-ITD have up to a 40% cure rate after HCT.3
In efforts to: 1) combat the downstream effects of an overexpressed or mutated FLT3 enzyme, especially with ITD; 2) minimize or overcome drug resistance, including that of FLT3 inhibitors; and 3) improve survival outcomes in AML, multiple research studies are being conducted with the ultimate goal of identifying safe and effective FLT3 inhibitors that could be utilized as monotherapy or in combination with standard (e.g., cytarabine, daunorubicin, idarubicin, azacitidine, decitabine) chemotherapies.1-9
Epidemiology and Etiology
AML is the most frequently occurring form of leukemia, with the highest incidence rates reported in the United States, Australia, and Western Europe.10 It accounts for about a quarter of all cases of adult leukemia in the Western world.10 AML predominantly affects patients who are in their sixth decade of life or older, with an average age of 66 years at diagnosis.11 The lifetime risk of AML for men is about 1 in 227, while it is about 1 in 278 for women (i.e., men are more likely to be diagnosed with AML).11 There is a 5-year relative survival rate of 24% with AML, which has slightly improved from the 21.7% observed between 1996 and 2002.10,12 The American Cancer Society estimates that about 18,860 new cases and 10,460 deaths from AML will occur this year in the U.S.11
There are numerous, identifiable risk factors for developing AML. These include, but are not limited to, cigarette smoking (up to 25% increased risk of AML), hazardous chemicals (e.g., benzene, formaldehyde), prior chemotherapy (e.g., alkylating agents, topoisomerase II inhibitors), radiation (depending on the type and area of exposure), other blood abnormalities (e.g., myelodysplastic syndromes, which have a poor prognosis), congenital syndromes (e.g., Down syndrome, trisomy 8), family history, and male gender.11-17
Current Management
There have been several developments in the risk stratification (i.e., the probability of treatment-related mortality [TRM] and resistance to standard therapy despite no TRM) and treatment of patients with AML.3 Of note, therapeutic failure in AML is due more to treatment resistance than to TRM.3 There are multiple treatment strategies for AML, including regimens for induction, consolidation, maintenance, and relapsed/refractory disease.18 Notably, the current standard of care for induction therapy is cytarabine plus an anthracycline (e.g., idarubicin or daunorubicin).18
Among several guidelines for the treatment of AML, selected risk-adapted treatment recommendations were published in the American Journal of Hematology in 2013.3 FLT3 inhibitors were recommended to be used as part of induction therapy in patients who have the FLT3-ITD mutation.3 Postremission treatment recommendations included HCT from a matched sibling or unrelated donor, a clinical trial including quizartinib after HCT, IV cytarabine 1 g/m2 twice daily for 6 days, or a clinical trial for patients who are not HCT candidates.3
Anecdotally, the choice and sequencing of any chemotherapy varies greatly between institutions and realistically depends on medical oncologists’ experiences, as well as on patients’ prognostic markers and expectations.
Available FLT3 Inhibitors
Although all of the FLT3 inhibitors currently available on the market are not FDA-approved for use in AML, they have been utilized off-label due to their multitargeted (i.e., nonspecific) activity.19-21 Examples of these oral drugs include sorafenib, sunitinib, and ponatinib (TABLE 1).22-34
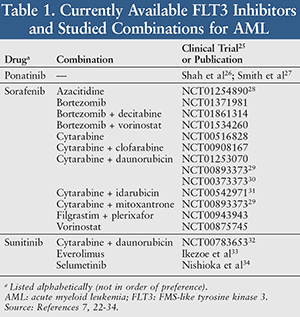
Sorafenib: Several phase I and II trials have included sorafenib in combination with other chemotherapy.25,28-31 Ravandi et al evaluated 51 newly diagnosed adult patients aged <65 years with AML, of which 13 had FLT3-ITD and two had FLT3-TKD mutations.31 Patients received sorafenib 400 mg twice daily (days 1-7) in combination with cytarabine 1.5 g/m2 IV daily (days 1-4) and idarubicin 12 mg/m2 IV daily (days 1-3). With a median follow-up of 54 weeks, there was an overall response rate (ORR) of 75%, median progression-free survival (PFS) of at least 60 weeks, and an overall survival (OS) of 83% at 6 months and 74% at 12 months. The combination was tolerated well, and grade 3 or higher adverse effects possibly related to adding sorafenib included increased liver function tests (LFTs; 5 patients), increased bilirubin (4), and diarrhea (4). The authors concluded that sorafenib could safely be combined with cytarabine and idarubicin, as well as induce high remission rates in adults <65 years of age with FLT3-mutated AML.31
The SORAML study evaluated 264 newly diagnosed adult patients <60 years of age.29 Patients received two cycles of “7+3” induction therapy with cytarabine 100 mg/m2 IV daily (days 1-7) and daunorubicin 60 mg/m2 IV daily (days 3-5). If these patients did not respond to the first cycle, they received “HAM” therapy with cytarabine 3 g/m2 IV twice daily (days 1-3) and mitoxantrone 10 mg/m2 IV daily (days 3-5). Patients also received sorafenib 400 mg twice daily or placebo (days 10-19). These patients then received three cycles of consolidation therapy with cytarabine 3 g/m2 IV twice daily (days 1, 3, and 5), along with sorafenib twice daily or placebo (day 8 until 3 days prior to the next consolidation cycle). They were then maintained on sorafenib twice daily or placebo for 12 months.29 With a median follow-up of 18 months, sorafenib and placebo were similar in complete response (CR; 60% vs. 56%; P = .622) and 2-year OS (72% vs. 66%; P = .367), but sorafenib had greater 1 year event-free survival (EFS; 64% vs. 50%; P = .023). Reported adverse effects that were grade 3 or higher included infectious complications (e.g., fever, pneumonia), bleeding events (relative risk of 3.6; P = .016), hepatotoxicity (relative risk of 6.2; P = .025), cardiotoxicity, hypertension, skin toxicity, and headache. The authors concluded that sorafenib can be added to standard chemotherapy in younger patients with AML, but it may lead to increased liver toxicity and bleeding issues.29
Serve et al evaluated sorafenib in combination with traditional chemotherapy in 197 newly diagnosed adult patients >60 years of age (median 68 y), of which 28 had a FLT3-ITD mutation.30 They received 7+3 induction therapy with cytarabine 100 mg/m2 IV daily (days 1-7) and daunorubicin 60 mg/m2 IV daily (days 3-5), followed by sorafenib 400 mg twice daily or placebo. Consolidation therapy included up to two cycles of cytarabine 1 g/m2 IV twice daily (days 1, 3, and 5), followed by sorafenib 400 mg twice daily or placebo. With a median follow-up of about 29 months, the median EFS (5 vs. 7 months), OS (13 vs. 15 months), ORR (57% vs. 64%), and CR (48% vs. 60%) were similar between sorafenib and placebo. Grade 3 or higher adverse events, which did not differ significantly between sorafenib and placebo, included febrile neutropenia, pneumonia, diarrhea, sepsis, skin rash, hemorrhage, mucositis, hypertension, cardiovascular events, high bilirubin, abdominal pain, arrhythmia, dyspnea, and hand-foot syndrome. The authors concluded that since there were no significant clinical differences between sorafenib and placebo, regardless of FLT3-ITD or NPM1 mutation status in patients, sorafenib combined with the aforementioned traditional chemotherapy is not recommended for patients aged >60 years.30
Ravandi et al also evaluated the combination of sorafenib and azacitidine in 43 patients who failed at least two regimens or relapsed with FLT3 inhibitor monotherapy or combination therapy.28 Patients had a median age of 64 years (range 24-87 y) and 40 had the FLT3-ITD mutation. They received sorafenib 400 mg twice daily (days 1-28) combined with azacitidine 75 mg/m2 IV or SC daily (days 1-7). With a median follow-up of 24 weeks and 37 evaluated patients, CR was 16%, ORR was 46%, median EFS was 3.8 months, and median OS was 6.2 months. Most patients experienced lower than grade 3 toxicities related to sorafenib, including fatigue and hepatotoxicity. Grade 3 or higher adverse events included thrombocytopenia, neutropenia (with fever or infection), anemia, and cardiomyopathy. Six deaths related to infection occurred during the treatment period. Gastrointestinal toxicity and myelosuppression led to azacitidine dose reductions in 4 patients. The authors concluded that sorafenib and azacitidine was an effective combination for patients with relapsed/refractory AML and FLT3-ITD.28
Sunitinib: In the AMLSG 10-07 trial, sunitinib combined with traditional chemotherapy was given to 22 adult patients >60 years of age, with a median age of 70 years (60-78 y).32 There were 15 patients with FLT3-ITD and 7 patients with FLT3-TKD mutations. Patients received up to two cycles of induction therapy with sunitinib 25 mg daily (days 1-7), cytarabine 100 mg/m2 IV daily (days 1-7), and daunorubicin 60 mg/m2 IV daily (days 1-3).32 They also received up to three cycles of consolidation therapy with sunitinib 25 mg daily (days 1-7) and cytarabine 1 g/m2 IV twice daily (days 1, 3, and 5). There was an ORR of 63.5%, CR of 53% in FLT3-ITD patients, and CR of 71% in FLT3-TKD patients. There were 7 patients on sunitinib maintenance therapy for a median of 11 months, with a median relapse-free survival of 11 months, median OS of 18.8 months, and 2-year survival of 36%. Major reported adverse events included myelosuppression, hand and foot syndrome, and colitis. The authors concluded that sunitinib could be combined with induction and consolidation chemotherapy.32
Ponatinib: Shah et al studied ponatinib monotherapy in 12 patients with relapsed or refractory AML who had a median age of 49 years (30-72 y).26 Seven patients had a FLT3-ITD mutation, but all received ponatinib 45 mg daily. With a median treatment duration of 52 days (10-173) and up to a 6-month follow-up, the ORR was 25% in all patients and 43% in FLT3-ITD patients who had not received any prior FLT3 inhibitor. The only grade-3 or higher toxicity was thrombocytopenia, but several other treatment-emergent adverse events occurred in at least 3 patients, including but not limited to febrile neutropenia, pneumonia, petechiae, peripheral edema, pyrexia, rash, pancreatitis, increased LFTs, lung infection, neoplasm progression, sepsis, intracranial hemorrhage, and leukocytosis. The authors concluded that despite the small sample size, ponatinib has clinical activity in patients with FLT3-ITD mutated AML.26
However, the FDA recently issued a warning about ponatinib, stating that the drug increases the risk of life-threatening blood clots and severe narrowing of blood vessels.35 Ponatinib is currently only available through an ARIAD PASS (Patient Access and Support Services) program, as part of a Risk Evaluation and Mitigation Strategy (REMS).36
Investigational FLT3 Inhibitors
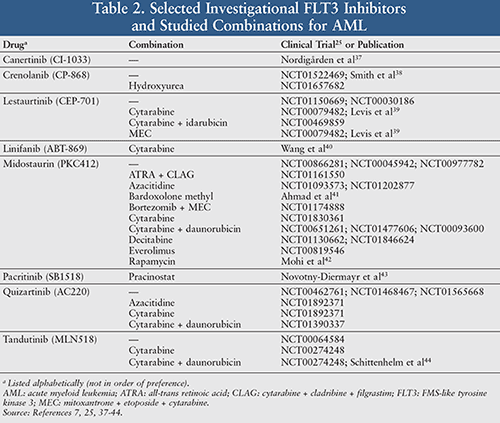
There are multiple FLT3 inhibitors currently under investigation, but most have yet to be evaluated in humans. TABLE 2 summarizes selected investigational FLT3 inhibitors, as well as studied combinations with other chemotherapies.25,37-44
Drug Resistance
While each FLT3 inhibitor listed in TABLEs 1 and 2 has unique pharmacokinetic and pharmacodynamic properties, the clinical efficacies of selected drugs (e.g., lestaurtinib, midostaurin, ponatinib, quizartinib, sorafenib, sunitinib, tandutinib) have not been satisfactory.7 FLT3-ITD cells can acquire TKD mutations (i.e., double mutants) following genotoxic chemotherapy, become less sensitive to FLT3 inhibitors (and continue proliferating regardless of FLT3 inhibition), and ultimately lead to disease progression.7
Since FLT3-ITD AML is also resistant to the most common “backbone” drug (i.e., cytarabine), it often warrants experimental combination uses of drugs that target other pathways, such as those depicted in FIGURE 1.7,8 Another potential strategy for overcoming resistance is utilizing novel multikinase inhibitors that target FLT3-ITD and -TKD (e.g., crenolanib), along with P1M1 (e.g., AR00459339), aurora kinase (e.g., CCT137690), and/or JAK2 (e.g., pacritinib, TG02).7,38,43-48
Conclusion
Specific FLT3 inhibition is a rational approach to treating AML patients, but its utility is currently limited due to disease heterogeneity and drug resistance.7,45 Most FLT3 inhibitors target the ITD mutation, but may eventually lead to dual FLT3-ITD and -TKD mutations that confer greater drug resistance.7 The prognostic factor of a TKD mutation itself may be unclear, but the combination of FLT3-ITD and -TKD mutations (along with AML cells escaping the FLT3 pathway itself) is of significant concern and is expected to lead to worse clinical outcomes for patients.7 Multifarious strategies that inhibit the mutated FLT3 enzyme and its downstream signaling pathways (e.g., aurora kinase, ERK, JAK2, mTOR, PIM1) may demonstrate enhanced efficacy against FLT3-mutated AML in future trials.7,45
ACKNOWLEDGMENT: The authors wish to acknowledge Nino Marzella, BS, MS, PharmD, and Shyamala C. Navada, MD, MSCR, for their professional guidance and support.
REFERENCES
1. Hasserjian RP. Acute myeloid leukemia: advances in diagnosis and classification. Int J Lab Hematol. 2013;35(3):358-366.
2. National Cancer Institute at the National Institutes of Health. Adult acute myeloid leukemia treatment (PDQ). www.cancer.gov/cancertopics/pdq/treatment/adultAML/Patient. Accessed October 12, 2014.
3. Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88(4):318-327.4. Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381(9865):484-495.
5. Alvarado Y, Kantarjian HM, Luthra R, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer. 2014;120(14):2142-2149.
6. Licht JD, Sternberg DW. The molecular pathology of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2005:137-142.
7. Leung AY, Man CH, Kwong YL. FLT3 inhibition: a moving and evolving target in acute myeloid leukaemia. Leukemia. 2013;27(2):260-268.
8. Swords R, Freeman C, Giles F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia. 2012;26(10):2176-2185.
9. Pemmaraju N, Kantarjian H, Ravandi F, et al. FLT3 inhibitors in the treatment of acute myeloid leukemia: the start of an era? Cancer. 2011;117(15): 3293-3304.
10. Deschler B, Lübbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006;107(9):2099-2107.
11. American Cancer Society. Leukemia: acute myeloid (myelogenous) overview. www.cancer.org/acs/groups/cid/documents/webcontent/003055-pdf.pdf. Accessed October 12, 2014.
12. American Cancer Society. Cancer Facts & Figures 2014. www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf. Accessed October 12, 2014.
13. Fircanis S, Merriam P, Khan N, Castillo JJ. The relation between cigarette smoking and risk of acute myeloid leukemia: an updated meta-analysis of epidemiological studies. Am J Hematol. 2014;89(8):E125-E132.14. Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol. 2008;35(4):418-429.
15. Pagano L, Pulsoni A, Vignetti M, et al. Secondary acute myeloid leukaemia: results of conventional treatments. Experience of GIMEMA trials. Ann Oncol. 2005;16(2):228-233.
16. Nardi V, Winkfield KM, Ok CY, et al. Acute myeloid leukemia and myelodysplastic syndromes after radiation therapy are similar to de novo disease and differ from other therapy-related myeloid neoplasms. J Clin Oncol. 2012;30(19):2340-2347.
17. Shi J, Shao ZH, Liu H, et al. Transformation of myelodysplastic syndromes into acute myeloid leukemias. Chin Med J (Engl). 2004;117(7):963-967.
18. Acute myeloid leukemia. HemOnc.org. http://hemonc.org/Acute_myeloid_leukemia. Accessed October 12, 2014.
19. Kayser S, Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2014;55(2):243-255.
20. Grunwald MR, Levis MJ. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol. 2013;97(6):683-694.
21. Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089-5102.
22. Nexavar (sorafenib oral tablets) package insert. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; November 2013.
23. Sutent (sunitinib malate oral capsules) package insert. New York, NY: Pfizer Inc; August 2013.
24. Iclusig (ponatinib oral tablets) package insert. Cambridge, MA: Ariad Pharmaceuticals, Inc; January 2014.
25. U.S. National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed October 12, 2014.
26. Shah NP, Talpaz M, Deininger MW, et al. Ponatinib in patients with refractory acute myeloid leukaemia: findings from a phase 1 study. Br J Haematol. 2013;162(4):548-52.
27. Smith CC, Lasater EA, Zhu X, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121(16):3165-3171.
28. Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655-4662.
29. Rollig C, Muller-Tidow C, Huttmann A, et al. Sorafenib versus placebo in addition to standard therapy in adult patients <60 years with newly diagnosed acute myeloid leukemia: results from the randomized-controlled SORAML trial. Presented at: the 54th American Society of Hematology Annual Meeting and Exposition; Atlanta, GA; December 9, 2012.
30. Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110-3118.
31. Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856-1862.
32. Sunitinib and intensive chemotherapy in patients with acute myeloid leukemia and activating FLT3 mutations: results of the AMLSG 10-07 study. Presented at: the 54th American Society of Hematology Annual Meeting and Exposition; Atlanta, GA; December 8, 2012.
33. Ikezoe T, Nishioka C, Tasaka T, et al. The antitumor effects of sunitinib (formerly SU11248) against a variety of human hematologic malignancies: enhancement of growth inhibition via inhibition of mammalian target of rapamycin signaling. Mol Cancer Ther. 2006;5(10):2522-2530.
34. Nishioka C, Ikezoe T, Yang J, et al. Blockade of MEK/ERK signaling enhances sunitinib-induced growth inhibition and apoptosis of leukemia cells possessing activating mutations of the FLT3 gene. Leuk Res. 2008;32(6):865-872.
35. FDA Drug Safety Communication: FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. www.fda.gov/Drugs/DrugSafety/ucm379554.htm. Accessed October 12, 2014.
36. ARIAD Pharmaceuticals, Inc. ARIAD PASS. www.ariadpass.com. Accessed October 12, 2014.
37. Nordigården A, Zetterblad J, Trinks C, et al. Irreversible pan-ERBB inhibitor canertinib elicits anti-leukaemic effects and induces the regression of FLT3-ITD transformed cells in mice. Br J Haematol. 2011;155(2):198-208.
38. Smith CC, Lasater EA, Lin KC, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A. 2014;111(14):5319-5324.
39. Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294-3301.
40. Wang ES, Yee K, Koh LP, et al. Phase 1 trial of linifanib (ABT-869) in patients with refractory or relapsed acute myeloid leukemia. Leuk Lymphoma. 2012;53(8):1543-1551.
41. Ahmad R, Liu S, Weisberg E, et al. Combining the FLT3 inhibitor PKC412 and the triterpenoid CDDO-Me synergistically induces apoptosis in acute myeloid leukemia with the internal tandem duplication mutation. Mol Cancer Res. 2010;8(7):986-993.42. Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101(9):3130-3135.
43. Novotny-Diermayr V, Hart S, Goh KC, et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J. 2012;2(5):e69.
44. Schittenhelm MM, Kampa KM, Yee KW, et al. The FLT3 inhibitor tandutinib (formerly MLN518) has sequence-independent synergistic effects with cytarabine and daunorubicin. Cell Cycle. 2009;8(16):2621-2630.
45. Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65-77.
46. Fathi AT, Arowojolu O, Swinnen I, et al. A potential therapeutic target for FLT3-ITD AML: PIM1 kinase. Leuk Res. 2012;36(2):224-231.
47. Hatzimichael E, Georgiou G, Benetatos L, Briasoulis E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am J Blood Res. 2013;3(1):29-51.
48. Poulsen A, William A, Blanchard S, et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J Mol Model. 2013;19(1):119-130.
To comment on this article, contact rdavidson@uspharmacist.com.


