US Pharm. 2012;37(3)(Oncology suppl):3-7.
ABSTRACT: Acquired hemophilia A (AHA) is a rare autoimmune disorder whose incidence increases with age; its occurrence is uncommon in children aged <16 years. Morbidity and mortality are high, primarily because of bleeding and adverse effects of immune therapy. Treatment involves managing acute bleeding episodes and eradicating inhibitors. Lack of familiarity with AHA can result in delayed diagnosis and suboptimal treatment, so the patient should be referred to a hematology specialist who is experienced in treating the disorder.
Acquired hemophilia A (AHA) is a rare autoimmune disorder with an annual incidence of 1.5 cases per million.1 Unlike congenital hemophilia, AHA incidence increases with age, and cases are uncommon in children aged <16 years (0.045 cases/million/year). The incidence in individuals aged >85 years is 14.7 cases per million per year and likely is underestimated. Incidence is similar between men and women except for the age group of 20–40 years, in which the incidence is higher in women because of the increased risk conferred by pregnancy.2
AHA is characterized by the presence of autoantibodies against factor VIII (FVIII). The most common FVIII inhibitors in AHA are immunoglobulin G (IgG)1 and IgG4 autoantibodies. The antibodies interfere with the coagulant effect of FVIII.3 Morbidity and mortality are high in AHA, primarily because of bleeding and adverse effects of immune therapy.4 Bleeding severity ranges from severe to minor. The incidence of mortality from bleeding is reported to be as high as 31% and as low as 9%, with the latter incidence noted in more recent years as therapeutic options have evolved.2,5 Recently, the European Acquired Haemophilia Registry 2 (EACH2) reported a rate of 3% for fatal bleeding (which can occur ≤5 months after initial presentation if inhibitor eradication is unsuccessful).6 There is no correlation between inhibitor titer/FVIII levels and bleeding severity.7 An underlying medical condition (TABLE 1) is identified in up to 50% of AHA patients.3,7,8 The advanced age of many patients presenting with AHA often impacts clinical phenotype and treatment selection.

A hematologist experienced in inhibitor management should be sought regardless of the severity of clinical presentation. Lack of familiarity with AHA may lead to delayed diagnosis and suboptimal treatment. An interim report of the EACH2 registry found a median (5th-95th percentile) delay of 3 days (range 0-58 days) from bleeding onset to diagnosis. Additionally, a 1-day (range 0-69 days) difference between first abnormal activated prothrombin time (aPTT) and AHA diagnosis was noted.9 These data suggest a delay in AHA identification that puts patients at risk for bleeding.
The following review highlights the clinical presentation, diagnosis, and treatment of AHA. The acute management of bleeding and inhibitor eradication therapy are evaluated.
Diagnosis
Clinical Presentation: Any recent onset of bleeding with unexplained aPTT prolongation should raise suspicion of AHA, especially in the setting of advanced age or the peripartal period.1,3 The most common clinical presentation in AHA is subcutaneous bleeding and ecchymosis (80% of cases).4 In an analysis of 172 patients with AHA, gastrointestinal (23%), genitourinary (9%), retroperitoneal (9%), joint (7%), and intracranial (3%) hemorrhages were commonly reported.2 Mild bleeding not requiring hemostatic treatment occurs in up to 30% of cases.2 It is difficult to predict bleeding severity, as clinical phenotype does not correlate with FVIII levels or inhibitor titers.10 Advanced age and patient comorbidity often dictate the clinical presentation (bleeding risk and treatment-related adverse events).1
Laboratory Tests: Once a diagnosis of AHA is suspected, it should be confirmed by laboratory findings. Initial laboratory investigation in a patient with unexplained bleeding includes a CBC to assess platelet count and a coagulation panel. The typical finding in AHA patients is an isolated prolonged aPTT accompanied by a low FVIII level.1 Not all AHA patients present with significant aPTT prolongation or bleeding.7 Thrombin times and PTTs are normal, as are the platelet count and function. A prolonged aPTT accompanying a normal PTT may be due to a deficiency in one of the intrinsic coagulation factors (FVIII, IX, XI, or XII) or indicate the presence of an inhibitor.1 It is critical to distinguish between FVIII inhibitors and antiphospholipid antibodies (APAs) (lupus anticoagulant), as APAs may artificially lower FVIII, thereby mimicking AHA.7
Coagulation factor deficiencies, lupus anticoagulants, and heparin therapy may affect aPTT. To differentiate between a factor deficiency and the presence of an inhibitor, a mixing test is performed. In the test, plasma from the patient is mixed with pooled normal plasma in a 1:1 ratio. The premise is that if the patient has a prolonged aPTT caused by a factor deficiency, the pooled normal plasma will provide the missing factor, resulting in correction of the aPTT. Conversely, noncorrection of the aPTT suggests the presence of an inhibitor.1 Mixing studies should be conducted immediately before and after 2 hours of incubation since FVIII inhibitors are time- and temperature-dependent (incubation should be performed at 37°C).7 Failure of aPTT correction by more than 50% subsequent to mixing with normal plasma suggests the presence of an inhibitor; however, it is not definitive for AHA.11 A laboratory experienced in hemostasis should be consulted for further testing.
An additional test is the Bethesda assay, which was developed to quantify FVIII inhibition. Acquired inhibitors to FVIII display complex and nonlinear type 2 kinetics, whereas alloantibodies display linear type 1 kinetics.7 The Bethesda assay measures the concentration, or inhibitor titer, expressed in Bethesda units (BU/mL). One BU is defined as the titer of antibodies inactivating 50% of FVIII activity in a mixture of equal volumes of examined plasma and normal plasma after 2-hour incubation at 37°C.7
Treatment
Management of Acute Bleeding Episodes: Treatment of acute bleeding may be stratified by patient inhibitor level. Patients with a low inhibitor level (<5 BU) may be managed by increasing FVIII levels via FVIII concentrates (plasma-derived or recombinant). Increasing plasma FVIII levels by 30% to 50% typically achieves hemostasis; nonetheless, it is quite difficult to predict patient response. The optimal approach is to tailor treatment to the patient, utilizing past experience as a gauge.8 Although the optimal dosing of FVIII in AHA is yet to be defined, a dose of 200 IU/kg via bolus injection every 8 to 12 hours has been suggested.12 A dose of 20 IU/kg for each BU of inhibitor present, plus an additional 40 IU, has also been recommended. Ten to 15 minutes after the initial bolus, a plasma FVIII level should be drawn. If patient response is inadequate, another bolus should be given.10 Ultimately, therapy must be closely monitored and adjusted according to plasma FVIII levels and patient response.
In patients with very low inhibitor titers <3 BU, an IV infusion of desmopressin 0.3 mcg/kg may provide benefit by increasing FVIII and von Willebrand factor levels by two- to threefold. Use of desmopressin in patients with higher titer levels is suboptimal and may delay therapy that utilizes more effective options.10
In patients with inhibitor levels ≥5 BU who present with an acute bleeding episode, the treatment of choice includes activated prothrombin complex concentrate (aPCC) (FEIBA; Baxter) or recombinant activated factor VIIa (rFVIIa) (NovoSeven; NovoNordisk).13 In patients presenting with acute bleeding episodes, recent guidelines recommend either aPCC 50-100 IU/kg via bolus injection every 8 to 12 hours (maximum 200 IU/kg/day) or rVIIa 90 mcg/kg every 2 to 3 hours until hemostasis.7 If the patient fails to respond to one agent, a viable option is to switch to the alternative agent. More than 80% of patients treated with either agent respond to treatment (TABLE 2).14-22 To date, there have been no head-to-head comparisons of these agents; however, each agent possesses advantages and disadvantages.
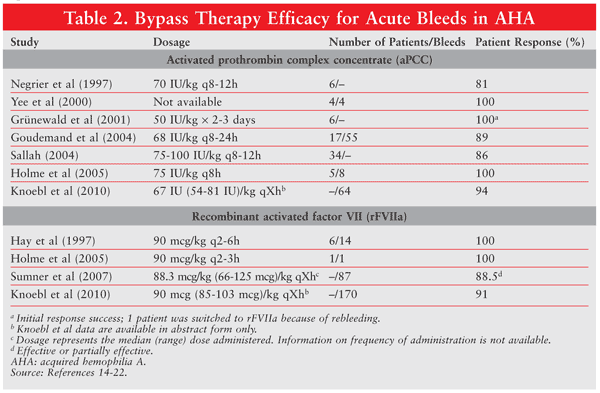
The primary disadvantages of rFVIIa are the frequent dosing (every 2-3 hours) and high cost. aPCC is plasma derived and has the potential for contamination. Precautions to minimize contamination with infectious agents have been taken, as aPCC is double virus–inactivated. Concerns over bloodborne illness are minimal with rFVIIa, since it is a recombinant protein. Regardless, no transmission of bloodborne illness has been documented with either agent. Both agents carry the risk (although rare) of venous thromboembolism, disseminated intravascular coagulation, and myocardial infarction.23 Sumner and colleagues reported a thrombotic event rate of 8.6% in 139 AHA patients treated with rFVIIa.21 Recently, an analysis of EACH2 registry data revealed a lower thrombotic event rate (2.3%) with hemostatic therapy.22 To mitigate the risk, the lowest effective dose should be used, and doses should not exceed manufacturer recommendations.
Aminocaproic acid and tranexamic acid have been reported to be effective in managing acute bleeding; however, no consensus or guideline is available on the appropriate dosage for and usage in AHA.24
Immune Therapy: Eradication of Inhibitors
Once AHA has been diagnosed, inhibitor eradication therapy should be initiated immediately.1,7,25 Even if bleeding (minor or major) is controlled, the patient remains at risk for life-threatening bleeding until the inhibitor has been eradicated.2 Several options for inhibitor eradication are supported in the literature, including corticosteroids, cyclophosphamide, azathioprine, vincristine, rituximab, cyclosporine, plasmapheresis, and FVIII tolerance.1,7
Although clinical studies and reports support the various strategies for inhibitor eradication, they are difficult to compare and interpret because of their heterogeneity. In interpreting these data, it should be remembered that spontaneous remission occurs in 36% of patients with hemophilia.5 This does not imply that delaying treatment in the hope of spontaneous remission is an option, since sequelae that could arise while response is awaited may produce significant morbidity or mortality; rather, the data suggest that treatments producing a response rate <36% should be carefully evaluated.
Corticosteroids/Corticosteroids Plus Cyclophosphamide and Other Cytotoxics: The most common regimens used for inhibitor eradication are prednisolone monotherapy (1 mg/kg/day) and prednisolone plus cyclophosphamide (1-2 mg/kg/day).1 Some experts suggest that combination therapy achieves inhibitor eradication more rapidly. To date, data have not supported—nor have consensus guidelines recommended—one regimen over the other.1 Both strategies have been noted to achieve remission in >60% of patients; however, some reports have suggested that remission is greater (80%) with the addition of cyclophosphamide.9 In general, with treatment, the median time to remission is approximately 5 weeks.4 If a reduction in inhibitor titer is not seen in 2 to 3 weeks, an alternative method should be considered.1 Disease-free survival and overall survival are similar between corticosteroid monotherapy and corticosteroid-plus-cyclophosphamide therapy.10,26
Alternative strategies include the addition of cyclophosphamide or rituximab and combinations of cytotoxic agents. Several other cytotoxics have been combined with corticosteroids, including azathioprine, vincristine, and mycophenolate mofetil.1,27,28 Azathioprine is the preferred cytotoxic in pregnant patients.26 The benefits of using these alternatives must be carefully weighed against the risk of toxicity, namely, myelosuppression. Severe neutropenia leading to infection and subsequent sepsis syndrome has been reported.1 Particular caution should be taken in elderly patients, as they often have comorbidities, which increase the risk of toxicity.
Rituximab: The administration of rituximab 375 mg/m2 once weekly for 4 weeks has demonstrated promising results. Several studies have reported complete remission (CR) in 80% to 100% of patients treated.29-31 The median time to response is 2 weeks.8 The data are confounded by the concomitant immunosuppressive agents administered in the majority of cases, making it difficult to determine rituximab’s contribution. Nonetheless, rituximab may be a viable option in patients who have failed first-line treatment or who cannot tolerate cytotoxics. Aggarwal and colleagues have suggested a treatment algorithm defining the role of rituximab: the addition of rituximab to prednisone in patients with inhibitor titers ≥5 BU but <30 BU, and to prednisone and cyclophosphamide in patients with titers ≥30 BU.32 Singh and colleagues evaluated durability of response to a rituximab-containing regimen in eight consecutive patients with AHA.33 The initial response rate was 88% (7/8 patients), and all responsive patients remained in CR after a median follow-up of 5 years. As with previous reports, most patients received other forms of immunosuppressants concomitantly with rituximab.
IV Immunoglobulin (IVIG): IVIG 2 g/kg over 2 to 5 days has been used for eradication. Schwartz and colleagues noted a response rate of 30% with this regimen.34 Patients who responded tended to have lower inhibitor titers, and maximal response was seen after a mean of 40 days. Conversely, a meta-analysis by Delgado and colleagues found no advantage to adding IVIG to therapy.10 Current guidelines recommend against the use of IVIG in AHA.7
Cyclosporine: To date, a limited number of cases have had CR with the use of cyclosporine.35,36 Cyclosporine may be particularly useful in patients with systemic lupus erythematosus.8,37 The dosage should be titrated to maintain a serum trough level of 200 to 400 mcg/mL.35,36,38 Case reports have indicated success with trough levels <200 mcg/mL.35,38 A typical initiation dose is 200 to 300 mg by mouth daily.37,38
Immunoadsorption: Autoantibodies to FVIII are removable via plasmapheresis or immunoadsorption using staphylococcal protein A.39 In addition to extracorporeal removal, patients should receive agents to augment FVIII levels.1 Although antibodies are removable, immunosuppressive agents should be added to the procedure since immunoadsorption has not been proven to shorten the time to CR.10
Immune Tolerance Induction (ITI): Nemes and Pitlik examined the effectiveness of the ITI Budapest protocol in patients with AHA.40 An aggressive 3-week protocol was used (week 1, human FVIII 30 U/kg/day; week 2, FVIII 20 U/kg/day; week 3, 15 U/kg/day; cyclophosphamide 200 mg/day to a total dose of 2-3 g; and methylprednisolone 100 mg/day IV for 1 week, then tapered over 2 weeks) to achieve inhibitor eradication in 13 of 14 patients receiving ITI.40 ITI is rarely used in adults; it is primarily used in young patients with congenital hemophilia and FVIII alloantibodies.10
Special Treatment Considerations
Treatment selection should be individualized in order to minimize toxicity. In a retrospective review of 172 patients with AHA, Delgado and colleagues concluded that approximately one-half of patients experienced morbidity unrelated to bleeding.41 Morbidity was more often related to adverse effects of immunosuppression. To minimize unwanted outcomes, it is crucial to implement the appropriate drug and to utilize a dosage that has been adjusted for comorbidity (i.e., renal and/or hepatic impairment).
In patients with underlying malignancy, inhibitor eradication should take place after the malignancy has been treated. Inhibitor eradication is more attainable once the primary malignancy is controlled.10
Alkylating agents such as cyclophosphamide carry the risk of infertility; therefore, postpartum women should not receive these agents.25 Azathioprine may be particularly useful in patients who are not candidates for alkylating agents.
Patients should be instructed that invasive diagnostic or therapeutic procedures are generally best avoided. Minor procedures such as venipuncture, peripheral catheter placement, and blood pressure measurement should be performed with caution, since they may cause severe bleeding events. Intramuscular injections are contraindicated.
Perioperative management is challenging in AHA. Procedures should be performed only if absolutely necessary and the benefits outweigh the risks. There are no strategies that guarantee hemostasis; however, data support both aPCC and rFVIIa as effective for prophylaxis management.20,42,43 Postponement of procedures until inhibitor eradication has been achieved should be considered, if possible.7
Conclusion
AHA is a rare disease that is associated with significant morbidity and mortality. The various available treatment approaches may be divided into two main categories: management of the acute bleeding episode and inhibitor eradication. Much of the literature supporting treatment is based on case series, case reports, and expert opinion. Given the paucity of data available for the optimal treatment of AHA, it is critical to enroll patients in national and international registries, which will help further understanding of the best treatment approach. Delayed diagnosis or inappropriate treatment may place patients at risk for significant morbidity or mortality. Patients should be referred to a hematology specialist experienced in the treatment of AHA.
REFERENCES
1. Collins P, Baudo F, Huth-Kühne A, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010;3:161.
2. Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in
the United Kingdom: a 2-year national surveillance study by the United
Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109:1870-1877.
3. Shetty S, Bhave M, Ghosh K. Acquired hemophilia A: diagnosis, aetiology, clinical spectrum and treatment options. Autoimmun Rev. 2011;10:311-316.
4. Collins PW. Treatment of acquired hemophilia A. J Thromb Haemost. 2007;5:893-900.
5. Lottenberg R, Kentro TB, Kitchens CS. Acquired hemophilia. A
natural history study of 16 patients with factor VIII inhibitors
receiving little or no therapy. Arch Intern Med. 1987;147:1077-1081.
6. Marco P, Collins P, Knoebl P, et al. Acquired haemophilia:
clinical and demographic data. Results of European Acquired Haemophilia
Registry (EACH2). Blood. 2010;116:abstract 1398.
7. Huth-Kühne A, Baudo F, Collins P, et al. International
recommendations on the diagnosis and treatment of patients with acquired
hemophilia A. Haematologica. 2009;94:566-575.
8. Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program. 2006:432-437.
9. Collins PW. Management of acquired haemophilia A. J Thromb Haemost. 2011;9(suppl 1):226-235.
10. Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A.
Acquired haemophilia: review and meta-analysis focused on therapy and
prognostic factors. Br J Haematol. 2003;121:21-35.
11. Kasper CK. Complications of hemophilia A treatment: factor VIII inhibitors. Ann N Y Acad Sci. 1991;614:97-105.
12. Kessler CM, Gill JC, White GC II, et al. B-domain deleted
recombinant factor VIII preparations are bioequivalent to a monoclonal
antibody purified plasma-derived factor VIII concentrate: a randomized,
three-way crossover study. Haemophilia. 2005;11:84-91.
13. Tjonnfjord GE, Holme PA. Factor eight inhibitor bypass activity
(FEIBA) in the management of bleeds in hemophilia patients with
high-titer inhibitors. Vasc Health Risk Manag. 2007;3:527-531.
14. Negrier C, Goudemand J, Sultan Y, et al. Multicenter
retrospective study on the utilization of FEIBA in France in patients
with factor VIII and factor IX inhibitors. French FEIBA Study Group.
Factor Eight Bypassing Activity. Thromb Haemost. 1997;77:1113-1119.
15. Yee TT, Taher A, Pasi KJ, Lee CA. A survey of patients with
acquired haemophilia in a haemophilia centre over a 28-year period. Clin Lab Haematol. 2000;22:275-278.
16. Grünewald M, Beneke H, Güthner C, et al. Acquired haemophilia: experiences with a standardized approach. Haemophilia. 2001;7:164-169.
17. Goudemand J, Tagariello G, Lopaciuk F. Cases of surgery in high-responder haemophilia patients. Haemophilia. 2004;10(suppl 2):46-49.
18. Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia. 2004;10:169-173.
19. Holme PA, Brosstad F, Tjonnfjord GE. Acquired haemophilia:
management of bleeds and immune therapy to eradicate autoantibodies. Haemophilia. 2005;11:510-515.
20. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in
acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost. 1997;78:1463-1467.
21. Sumner MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of
acquired haemophilia with recombinant activated FVII: a critical
appraisal. Haemophilia. 2007;13:451-461.
22. Knoebl P. Management of bleeding in acquired hemophilia: results of the European Acquired Hemophilia Registry (EACH2). Blood. 2010;166:abstract 716.
23. Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII
inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic
adverse events. Haemophilia. 2002;8:83-90.
24. Sahu S, Raipancholia R, Pardiwalla FK, Pathare AV. Hemostasis in
acquired hemophilia—role of intracavitary instillation of EACA. J Postgrad Med. 1996;42:88-90.
25. Holme PA, Glomstein A, Gronhaug S, Tjonnfjord GE. Home treatment
with bypassing products in inhibitor patients: a 7.5-year experience. Haemophilia. 2009;15:727-732.
26. Hay CR, Brown S, Collins PW, et al. The diagnosis and management
of factor VIII and IX inhibitors: a guideline from the United Kingdom
Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133:591-605.
27. Lian EC, Larcada AF, Chiu AY. Combination immunosuppressive
therapy after factor VIII infusion for acquired factor VIII inhibitor. Ann Intern Med. 1989;110:774-778.
28. Eisert S, Mosler K, Laws HJ, Göbel U. Successful use of
mycophenolate mofetil and prednisone in a 14-year-old girl with acquired
hemophilia A. Thromb Haemost. 2005;93:792-793.
29. Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell
depletion with rituximab for the treatment of patients with acquired
hemophilia. Blood. 2004;103:4424-4428.
30. Wiestner A, Cho HJ, Asch AS, et al. Rituximab in the treatment of acquired factor VIII inhibitors. Blood. 2002;100:3426-3428.
31. Onitilo AA, Skorupa A, Lal A, et al. Rituximab in the treatment of acquired factor VIII inhibitors. Thromb Haemost. 2006;96:84-87.
32. Aggarwal A, Grewal R, Green RJ, et al. Rituximab for autoimmune haemophilia: a proposed treatment algorithm. Haemophilia. 2005;11:13-19.
33. Singh AG, Hamarneh IS, Karwal MW, Lentz SR. Durable responses to rituximab in acquired factor VIII deficiency. Thromb Haemost. 2011;106:172-174.
34. Schwartz RS, Gabriel DA, Aledort LM, et al. A prospective study
of treatment of acquired (autoimmune) factor VIII inhibitors with
high-dose intravenous gammaglobulin. Blood. 1995;86:797-804.35. Au WY, Lam CC, Kwong YL. Successful treatment of acquired factor VIII inhibitor with cyclosporin. Haemophilia. 2004;10:98-100.
36. Pardos-Gea J, Ordi-Ros J, Altisent C, et al. Acquired haemophilia
A: successful treatment with immunosuppression, methylprednisolone
pulses and oral cyclosporin. Thromb Haemost. 2006;95:735-737.
37. Schulman S, Langevitz P, Livneh A, et al. Cyclosporine therapy
for acquired factor VIII inhibitor in a patient with systemic lupus
erythematosus. Thromb Haemost. 1996;76:344-346.
38. Petrovic M, Derom E, Baele G. Cyclosporine treatment of acquired hemophilia due to factor VIII antibodies. Haematologica. 2000;85:895-896.
39. Rivard GE, St Louis J, Lacroix S, et al. Immunoadsorption for
coagulation factor inhibitors: a retrospective critical appraisal of 10
consecutive cases from a single institution. Haemophilia. 2003;9:711-716.
40. Nemes L, Pitlik E. New protocol for immune tolerance induction in acquired hemophilia. Haematologica. 2000;85(suppl 10):64-68.
41. Delgado J, Villar A, Jimenez-Yuste V, et al. Acquired hemophilia:
a single-center survey with emphasis on immunotherapy and
treatment-related side-effects. Eur J Haematol. 2002;69:158-164.
42. Rodriguez-Merchan EC, Rocino A, Ewenstein B, et al. Consensus
perspectives on surgery in haemophilia patients with inhibitors: summary
statement. Haemophilia. 2004;10(suppl 2):50-52.
43. Tjonnfjord GE. Activated prothrombin complex concentrate (FEIBA)
treatment during surgery in patients with inhibitors to FVIII/IX: the
updated Norwegian experience. Haemophilia. 2004;10(suppl 2):41-55.
To comment on this article, contact rdavidson@uspharmacist.com.



