US
Pharm. 2006;5:39-48.
Systemic lupus erythematosus (SLE) is a chronic, multisystemic, inflammatory,
autoimmune disorder characterized by unpredictable exacerbations, remissions,
and immunologic manifestations.1 Approximately 1.5 million
Americans have a form of lupus, 70% of which is systemic.2 Although
lupus can affect both men and women at any age, 90% of individuals diagnosed
with the disease are women, and 80% of all patients with systemic lupus
develop it between ages 15 and 45.2 SLE is characterized by a
predilection for clinical involvement of the skin, joints, and almost any
organ and/or tissues of the body, including the heart, kidneys, blood, lungs,
and brain.2,3 Patients with SLE have a continual risk of clinical
exacerbation; in addition, more than half of these patients will develop
severe organ damage in various combinations.1,2,4
Background
SLE occurs 10 to 15 times more often in adult females than in adult males and
is most prevalent among women of childbearing age.1,2,5,6 As is the
case with gender, ethnicity also has a strong effect on SLE expression; the
disease occurs two to three times more often in African-Americans than in
Caucasians.1-5 African-Americans diagnosed with SLE are reported to
have an earlier onset, increased severity of disease, and earlier mortality.
5 Women of Hispanic, Asian, and Native American descent are also at
increased risk for SLE.6,7
While the prognosis of SLE has significantly improved, the mortality rate of
patients with SLE, who tend to be young or middle-aged, is at least three
times that of the general population.1,3 Nevertheless, survival
rates for SLE have improved, rising to approximately 95% at five years and 90%
at 10 years after diagnosis.3,6 The decrease in mortality can be
attributed to earlier diagnosis of disease, improvement in disease-specific
treatments, and advancements in general medical care. Mortality in patients
with long-standing SLE is often related to malignancy, renal, and
cardiovascular complications,whereas mortality in patients with recently
diagnosed SLE is usually related to active disease and infections.3,4,6
Pathophysiology
SLE is a complex autoimmune disease of unknown etiology. The disease is
characterized by immune dysregulation resulting in sustained production of
pathogenic autoantibodies, formation of circulating immune complexes, and
activation of the complement system.1,3-8 The production of
antinuclear antibodies (ANA) occurs in more than 95% of patients with SLE.
8 This dysregulation results in autoimmune reactions against host
antigens, which lead to inflammation and tissue damage that cause cellular and
organ dysfunction. Other autoantibodies, such as anti–double -stranded DNA
and anti-Smith, are also very specific for SLE.6,8,9
While the etiology of SLE remains obscure, environmental, genetic, and
hormonal factors have an important role in the pathogenesis of the disease.
8-11 Clinical flares and exacerbations occur in approximately 70% of
patients with SLE who are exposed to sunlight or ultraviolet light.8
Chemical, bacteria, or viral antigens in genetically predisposed individuals
may also trigger SLE. A strong familial predisposition has been suggested,
particularly among first-degree relatives.1,8 Hormonal factors can
also predispose individuals to the disease; women receiving hormone
replacements or estrogen-containing medications have an approximately twofold
increased risk of developing SLE.8,10
Clinical Manifestations
SLE is a chronic autoimmune disease that produces clinical manifestations and
inflammatory involvement of one or several organ systems.8,12 The
most common pattern is a mixture of constitutional symptoms with
musculoskeletal, skin, renal, and serologic involvement.6 Clinical
findings may include cardiac abnormalities, neurological abnormalities,
hemolytic anemia, poly arthralgia, and polyserositis.8,12 The
most common clinical symptoms are fever, rash, extreme fatigue, and arthritic
pain. While approximately 90% of all patients with SLE will present with
painful or swollen joints and arthralgias, 70% of patients will experience a
characteristic erythematous skin rash known as butterfly or malar
rash.7-9 About half of all patients with SLE develop
nephropathy, while 70% are photosensitive and complain of a photosensitivity
rash.7,8 Patients with SLE are at an increased risk for renal
dysfunction, cardiovascular disease, and infections of the respiratory and
urinary systems.3,6,8,12 Other clinical symptoms may include chest
pain, alopecia, oral ulcerations, headaches, ataxia, confusion, seizures, and
depression.6,12 Since the severity of symptoms varies greatly, the
course of the disease and common complications of SLE are best understood by
reviewing the major areas of potential disease involvement (table 1).1,6
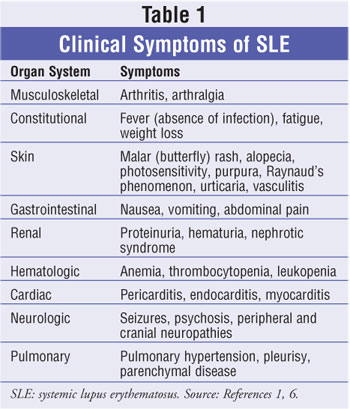
Drug-Induced Lupus Erythematosus
Drug-induced lupus erythematosus (DILE) is a syndrome that resembles SLE and
is associated with symptoms such as fever, malaise, arthritis, serositis,
and/or rash.8 DILE has a less severe and less dramatic clinical
presentation than SLE and occurs as a result of a hypersensitivity reaction
with certain medications and biologic agents. DILE has less female
predilection than SLE, is predominant in Caucasians, rarely involves the
kidneys or brain, and usually resolves within weeks or months after
discontinuation of the offending medication.8
Diagnosis
Due to the broad clinical and immunologic manifestations and varying symptoms
of SLE, the disease should be suspected in patients who present with clinical
symptoms affecting two or more of the organ systems listed in table 1.1
Diagnosis of SLE can be challenging because patients are prone to
unpredictable exacerbations, remissions, and conditions that can mimic disease
flares. Therefore, it is important to note that no single test can determine
whether a person has SLE. Diagnosis is based on characteristic clinical
features and laboratory criteria.6 The revised 1982 American
College of Rheumatology (ACR) criteria for classification of SLE requires the
presence of at least four of 11 conditions at any time during a patient's
medical history (table 2).13,14 The ACR classification criteria are
the most widely used to confirm and categorize patients with a diagnosis of
SLE. Since these criteria also classify patients for clinical research studies
and often lack sensitivity to milder forms of SLE, the ACR criteria may be
less accurate in patients with mild disease.6,15 Consequently, the
ACR criteria should not be used solely to exclude or confirm a diagnosis of
SLE.
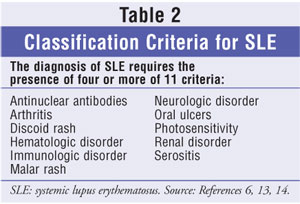
Diagnosis is confirmed by the presence of elevated ANA titers to 1:40 or higher.6,8 During the course of SLE, more than 95% of patients will have an elevated ANA titer. 6,8 Although several patients will have negative ANA titers early in disease, repeated negative tests suggest that the diagnosis is not SLE. The presence of ANA or multiple autoantibodies without clinical symptoms should not be considered diagnostic for SLE, although such persons are at increased risk.6,15 According to the ACR, patients in whom SLE is suspected based on history, characteristic signs and symptoms, or a positive ANA test should be referred to a rheumatologist to establish or confirm diagnosis.1
Treatment
Although there is no cure for SLE, advances in therapy over the past 50 years
have led to significant improvements in the life expectancy of patients with
SLE. The major challenges for managing patients with SLE include controlling
severe disease flares, preventing organ damage, and developing maintenance
strategies that suppress symptoms to an acceptable level.8 It is
important to note that treatment for active SLE differs depending on disease
severity and organ systems involved.9,12 Treatment for SLE is
largely determined by individual disease manifestations and organ involvement
and often includes a combination of drugs. The goals of therapy depend on the
prevention of disease complications and its treatments, and on whether disease
manifestations are potentially reversible, life-threatening, or likely to
cause permanent organ dysfunction.1,8 Several medications
effectively treat specific manifestations of SLE. Therapies and their
respective doses are listed in table 3.
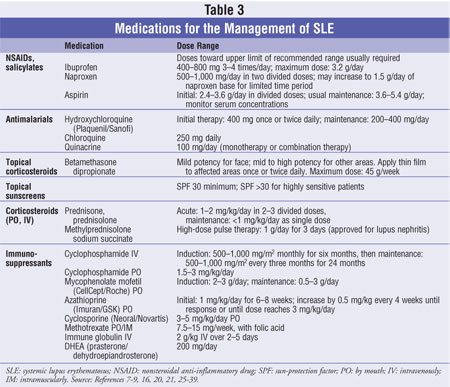
NSAIDs: More than
90% of patients with SLE will present with polyarthralgias or polyarthritis,
depending on the severity of the disease.8,9 NSAIDs such as
ibuprofen, which are effective analgesics/anti-inflammatories particularly for
arthritis and arthralgia, remain the mainstay of treatment for SLE patients.
NSAIDs also provide control of fever and symptomatic relief of headaches and
mild serositis. Compared to individuals without the disease, patients with SLE
have an increased risk of gastrointestinal (GI) toxicity, elevated serum
transaminases, hypertension, peripheral edema, and renal dysfunction.
Approximately half of all patients with SLE will develop associated nephritis.
In addition, it has been noted that NSAIDs can cause neuropsychiatric signs
and symptoms, aseptic meningitis (especially with ibuprofen use), and
salicylate-induced hepatitis.1,8,16 However, the most common side
effects associated with NSAID use are gastritis, gastric ulceration, and GI
bleeding.9 According to the ACR, patients at high risk for these
complications should be treated with gastroprotective agents such as H2
blockers, proton pump inhibitors, or prostaglandin analogs.1
Cyclooxygenase-2 (COX-2)
selective inhibitors: Recent data from clinical studies have raised
concern over potential increased health risks associated with the use of COX-2
selective inhibitors, such as celecoxib (Celebrex/Pfizer).1 Several
controlled clinical trials have demonstrated an increased risk of thrombotic
cardiovascular events with COX-2 selective inhibitors, particularly when used
at higher doses, longer durations of therapy, and in high-risk individuals.
1,17 Although these results also show that low doses (200 mg/day) of
celecoxib do not appear to be associated with increased risks, the majority of
these trials were at limited exposure ranging from approximately four to 12
weeks.17,18 According to the ACR, physicians and patients should
weigh the potential risks and benefits of these medications for the treatment
of SLE.1
Antimalarials
(hydroxychloroquine, chloroquine, and quinacrine): Antimalarial agents
are useful for the management of skin and joint manifestations, treatment of
constitutional symptoms (e.g., fever, fatigue, malaise), and prevention of
flares in SLE.9,16 Antimalarials often reduce dermatitis and
arthritis and have been shown to decrease levels of low-density lipoproteins.
8,16 Hydroxy chloroquine (Plaquenil/Sanofi) is the most widely used
antimalarial to treat SLE and may be used alone or in combination with other
drugs. Its effectiveness has also been studied in combination with quinacrine,
which can be added without increasing the risk of retinopathy.19
Although there have been reports of potential retinal toxicity with the use of
hydroxychloroquine, the risk is low; only 1% of patients using
hydroxychloroquine develop retinopathy.9,16 As a precaution, it is
recommended that patients receive an ophthalmologic examination before
initiation of therapy and at least every six to 12 months during therapy.
16 Adverse effects of antimalarial therapy include abdominal symptoms
(i.e., pain or dyspepsia), rashes or darkening of the skin, and muscle
weakness.
Corticosteroids
Topical or intralesional: Corticosteroids are naturally occurring
hormones with very potent anti-inflammatory properties. Many patients require
corticosteroids, alone or in combination with NSAID and antimalarial therapy,
to help control symptoms of SLE.9 Preparation of corticosteroids
include oral, intravenous (IV), topical, and intraarticular injections.
Topical or intralesional corticosteroids, such as betamethasone dipropionate,
are recommended for patients with localized cutaneous manifestations (e.g.,
malar rash, patchy erythema).1,7 Intralesional injections can be
used for discoid lesions, while intramuscular injections can be used to manage
generalized manifestations. The selection of corticosteroids depends on
current disease activity and severity. Fluorinated topical agents should not
be used for more than two weeks because of the potential development of
epidermal atrophy, depigmentation, and acne.7 Photosensitive
patients should be counseled to avoid sun exposure and to use sunscreen daily
with a sun-protection factor of 30 or higher.20,21 Sunscreen should
be applied 30 to 60 minutes prior to exposure and reapplied every four to six
hours.21
Systemic (oral and IV): The mainstay of SLE treatment for
life-threatening or multisystemic organ-threatening manifestations is systemic
corticosteroids, such as prednisone, hydrocortisone, methylprednisolone, and
dexamethasone.8,20 Systemic corticosteroids are used to control
severe disease flares in patients with SLE when NSAIDs, antimalarial agents,
and methotrexate are ineffective.9 Currently, high-dose IV
corticosteroids are used for refractory manifestations of SLE and are
recommended for shorter durations.8,16 Based on studies in lupus
nephritis, it has become standard practice to initiate therapy for
life-threatening SLE with pulses of high-dose IV corticosteroids.22
Prospective controlled trials in active lupus nephritis revealed that
administration of high doses of corticosteroids (methylprednisolone 1,000 mg
IV daily for three days) decreases the time to maximal improvements, as
compared to daily oral routes and cyclophosphamide.6,8,22 Dosages
should be tapered gradually once disease activity is under control.8
Low-dose prednisone (<=10 mg) is a reasonable maintenance dose; however,
the potential for toxicity is a concern.1,8,16
There are many complications associated with corticosteroid treatment,
including increased risk of infection, hyperglycemia, hypertension, and
osteoporosis.1,15 Electrolyte, glucose, and lipid levels should be
monitored in patients receiving long-term corticosteroid therapy to identify
metabolic complications. Patients should also receive bone densitometry to
identify osteoporosis and to monitor its response to treatment.23
All patients taking corticosteroids should be advised to take 1,000 mg daily
of supplemental calcium (1,500 mg for postmenopausal women) and 400 to 800 mg
of vitamin D, to minimize the risk of osteoporosis.23,24 Short-term
adverse effects associated with corticosteroid use include swelling, increased
appetite, weight gain, thinning of the hair, bruising, and dyspepsia.
1,7,15,23 These adverse effects generally cease once the drug is
discontinued.
Immunosuppressive/Cytotoxic
Medications
Immunosuppressive or cytotoxic medications are a class of drugs commonly used
for the treatment of severe, life-threatening SLE.1 These
medications have been researched extensively in patients with SLE who have
some manifestation of lupus nephritis.8,9 The treatment of SLE and
lupus nephritis can be divided into two phases: induction and maintenance. The
primary goal of induction immunosuppressive therapy is to induce immunologic
remission of the inflammatory manifestations of SLE, resulting in control of
the renal, extrarenal, and serologic signs of SLE. Once remission is achieved,
maintenance therapy is given for a prolonged period to help prevent relapse
and nonimmunologic progression of renal disease.8,12,25 Cytotoxic
medications may also induce a response to nonrenal manifestations of SLE such
as central nervous system manifestations, cytopenia, vasculitis, and pulmonary
hemorrhage.12 SLE patients receiving cytotoxic medications should
be monitored carefully for evidence of renal, hepatic, and hematologic
toxicity, as well as possible infection.1,12
Cyclophosphamide: Cyclophosphamide is the standard drug used for
serious, life-threatening active lupus nephritis. The use of cyclophosphamide
for the treatment of SLE has been reported successful in several studies when
used in combination with corticosteroid therapy.8,25,26 Responses
to cyclophosphamide begin within three to 16 weeks of treatment initiation,
whereas corticosteroid responses may begin within one day.8 The
recommended duration of cyclophosphamide therapy is controversial; however,
cyclophosphamide at an induction dose of 500 to 1,000 mg/m2 monthly
for six months is well tolerated when given in conjunction with prehydration
and mesna to avoid hemorrhagic cystitis.9,25,26 Adverse effects
commonly associated with cyclophosphamide use include myelosuppression,
premature ovarian failure, development of malignancies, nausea, malaise,
alopecia, and increased risk of infection.2,9,25
Azathioprine: Azathioprine (Imuran/GSK), an immunosuppressant and
purine antagonist used primarily as adjunct therapy to corticosteroids in the
treatment of SLE, is a less toxic alternative to cyclophosphamide for treating
nephritis.8,16 It decreases proliferation of immune cells,
resulting in lower autoimmune activity.27 Azathioprine reduces the
number of SLE flares and can be used as a steroid-sparing agent in nonrenal
disease.27 The agent may increase the risk of neoplasia,
pancreatitis, hepatotoxicity, and hematologic toxicities.1
Mycophenolate mofetil: Mycophenolate mofetil (CellCept/Roche) is
effective for managing both renal and nonrenal symptoms of SLE.28,29
Research has shown that the drug is well tolerated and has fewer side effects
than many other medications used to treat SLE.30 Mycophenolate
mofetil is also an effective cytotoxic agent in patients with severe SLE;
recent studies have shown vast improvement in patient outcomes with use of the
drug.28,30
Adverse reactions associated with the administration of mycophenolate mofetil
include constipation, diarrhea, nausea, vomiting, headache, abdominal pain,
leukopenia, sepsis, and an increased risk of infections.30
Cyclosporine: Cyclosporine (Neoral/Novartis) has also been used for
the treatment of severe, life-threatening SLE. Cyclosporine inhibits
production of interleukin-2 and inhibits T-lymphocyte functions. Cyclosporine
has a reported low efficacy-to-toxicity ratio in the treatment of SLE but is
nonetheless used by clinicians. Since cyclosporine is potentially nephrotoxic,
the doses used are relatively low (3 to 5 mg/kg/day orally) in patients
with steroid-resistant cytopenias of SLE or in patients with steroid
resistance who have developed bone marrow suppression from standard cytotoxic
agents.26 The principal adverse reactions associated with
cyclosporine therapy are renal dysfunction, tremor, hirsutism, hypertension,
and gum hyperplasia.
Methotrexate:
Methotrexate, a disease-modifying anti rheumatic drug, may be used in
patients to help control symptoms of active SLE. Methotrexate is used
primarily to manage arthritis, skin rashes below the neck that are refractory
to topical corticosteroids, serositis, and constitutional signs and symptoms
of SLE.8,9,16 Weekly, low-dose oral methotrexate (7.5 to 15 mg) can
be especially useful in addressing inflammatory arthritis and rashes
unresponsive to topical treatment. Methotrexate should be used with caution in
patients with lupus nephritis, renal impairment, or inflammation, because it
is eliminated by both glomerular filtration and tubular secretion.16
The most frequently reported adverse reactions with methotrexate include
mouth sores, ulcerative stomatitis, leukopenia, nausea, malaise, fatigue,
chills and fever, dizziness, headache, itching, skin rash, hair loss, and
decreased resistance to infection. Methotrexate use is also associated with
severe lung, bone marrow, and liver toxicity, including pneumonitis and
cirrhosis.1 Patients should be counseled to contact their physician
immediately if they experience difficulty breathing, wheezing, chest
tightness, unusual dry, persistent, nonproductive cough, fever, chills, or
swelling of face, lips, tongue, or throat.9,16,28
Immune globulin (IV): Immune globulin IV is used for the
immunosuppression of life-threatening SLE flares. Gamma globulin 2 g/kg IV for
two to five days has also produced short-term improvement in patients with
SLE-related immune thrombocytopenia or hemolytic anemia.31 In the
treatment of SLE, IV gamma globulin blocks the complement cascade, neutralizes
circulating myelin antibodies, and downregulates proinflammatory cytokines.
26,31 IV infusions may increase the risk of migraine attacks, aseptic
meningitis, urticaria, pruritus, thromboembolic events, volume depletion,
preexisting kidney disease, and renal tubular necrosis in elderly patients and
in patients with diabetes.31
Other Therapies
Dapsone and retinoids: For patients who cannot tolerate antimalarials
or who have complicated manifestations of SLE, dapsone and retinoids are
additional treatment options.9 These agents have been used
primarily to treat refractory skin lesions. Dapsone is a sulfone and may
produce toxic effects, which most commonly include destruction of red blood
cells and methemoglobinemia.21,32 It is important to check a
patient's glucose-6-phosphate dehydrogenase status before initiation of
dapsone therapy. Other effects include loss of appetite, nausea, rash,
nervousness, and peripheral neuropathy. Retinoids should not be used in
patients who may become pregnant.21
Prasterone/dehydroepiandrosterone (DHEA): The FDA recently approved an
expanded indication for orphan drug DHEA, allowing its use for the prevention
of loss of bone mineral density in patients with SLE who are receiving
corticosteroid therapy.33 Chemically similar to estrogen and
androgen and a precursor to both, DHEA is a natural steroid hormone found in
both sexes and is produced by the adrenal glands in greatest amounts during
early adulthood.34,35 Target organs convert the sulfate ester to
DHEA, where it is metabolized to androstenedione, the major precursor to
androgens and estrogens. DHEA is also believed to have immunomodulatory
effects that help correct defective interleukin-2 production in T lymphocytes
in patients with SLE. Studies have shown improvements in both bone density and
markers for bone resorption due to stimulation of bone formation, which is
attributed to the adrenergic properties of DHEA.33,34 Recent
studies have shown prasterone to be safe and effective for stabilizing active
disease flares and alleviating symptoms of SLE in women.33-35
Prasterone has also shown reductions in total cholesterol and triglyceride
levels.34 Adverse events related to use of prasterone include acne,
facial hair growth, and hormonal changes.
Rituximab: A primary goal of SLE therapy is preventing complications
of disease and its treatments. Several new biologic agents and selective
cytotoxic medications to treat SLE are in various phases of clinical trials in
the United States.36 Most treatment strategies disrupt the
production of T or B lymphocytes, particularly those undergoing activation.
37,38 Monoclonal anti–B lymphocyte antibodies may be beneficial for
patients with disease resistant to other immunosuppressive therapies.36
Clinical trials are currently testing the safety and effectiveness of
rituximab (Rituxan/Genentech and IDEC Pharmaceuticals), a monoclonal anti-CD20
antibody that blocks the production of B cells. According to recent studies,
the use of rituximab without cyclophosphamide may have some beneficial effects
but does not appear to reduce serologic markers of disease activity.36-38
Follow-up
Patients diagnosed with SLE require lifelong monitoring with physician
follow-up visits at least every three to six months, including a complete
patient history, physical examination, complete blood count, comprehensive
metabolic panel, hepatitis function panel, lipid panel,
urinalysis, and a 24-hour urine collection for protein.1 Patients
with more severe disease or starting immunosuppressant therapy will require
more frequent monitoring and follow-up.1
Conclusion
SLE is a chronic, complex, autoimmune disorder characterized by unpredictable
exacerbations, remissions, and immunologic manifestations. There is no cure
for SLE, but advances in therapy have led to significant improvements in
prognosis, survival, and patient outcomes. The major challenges for managing
patients with SLE include early diagnosis, appropriate referrals, prevention
of organ damage, and the use of treatment strategies that suppress symptoms.
Since SLE varies among patients, treatment includes a combination of
medications and is often determined by individual disease manifestations and
organ involvement. As the range of medications and effective treatments for
SLE has increased significantly, hope for advances in therapy and improvements
in quality of life continues.
REFERENCES
1. Guidelines for referral and management of systemic lupus erythematosus in
adults. American College of Rheumatology Ad Hoc Committee on Systemic Lupus
Erythematosus Guidelines. Arthritis Rheum. 1999;42:1785-1796.
2. Lahita RG. National Lupus Foundation of America. Education: Statistics
about Lupus. Available at: www.lupus.org/education/stats.html. Accessed
September 14, 2005.
3. Cervera R, Khamashta MA, Font J, et al. Morbidity and mortality in systemic
lupus erythematosus during a 10-year period: a comparison of early and late
manifestations in a cohort of 1,000 patients. Medicine (Baltimore).
2003;82:299-308.
4. Rivest C, Lew RA, Welsing PM, et al. Association between clinical factors,
socioeconomic status, and organ damage in recent onset systemic lupus
erythematosus. J Rheumatol. 2000;27:680-684.
5. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of
arthritis and selected musculoskeletal disorders in the United States.
Arthritis Rheum. 1998;41:778-799.
6. Gill JM, Quisel AM, Rocca PV, Walters DT. Diagnosis of systemic lupus
erythematosus. Am Fam Physician. 2003;68:2179-2186.
7. Wallace DJ, Metzger AL. Systemic lupus erythematosus: clinical aspects and
treatment. In: Koopman WJ, ed. Arthritis and Allied Conditions, 13th ed.
Baltimore: Williams and Wilkins; 1997:1319-1345.
8. Hahn BH. Systemic lupus erythematosus. In: Kasper D, ed. Harrison's
Principles of Internal Medicine. 16th ed. New York: McGraw-Hill;
2005:1960-1967.
9. Petri M. Treatment of systemic lupus erythematosus: an update. Am Fam
Physician. 1998;57:2753-2760.
10. Cooper GS, Dooley MA, Treadwell EL, et al. Hormonal, environmental, and
infectious risk factors for developing systemic lupus erythematosus. Arthritis
Rheum. 1998;41:1714-1724.
11. Pisetsky DS. Systemic lupus erythematosus: epidemiology, pathology, and
pathogenesis. In: Klippel JH, ed. Primer on the Rheumatic Diseases. 11th ed.
Atlanta: Arthritis Foundation; 1997:246-250.
12. Edworthy SM. Clinical manifestations of systemic lupus erythematosus. In:
Ruddy S, Harris ED, et al, eds. Kelley's Textbook of Rheumatology. 6th ed.
Philadelphia: Saunders; 2001:1105-1119.
13. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the
classification of systemic lupus erythematosus. Arthritis Rheum.
1982;25:1271-1277.
14. Hochberg MC. Updating the American College of Rheumatology revised
criteria for the classification of systemic lupus erythematosus [Letter].
Arthritis Rheum. 1997;40:1725.
15. Pisetsky DS, Gilkeson G, St. Clair EW. Systemic lupus erythematosus:
diagnosis and treatment. Med Clin North Am. 1997;81:113-127.
16. Klippel JH. Systemic lupus erythematosus treatment. In: Klippel JH, ed.
Primer on the Rheumatic Diseases. 11th ed. Atlanta: Arthritis Foundation;
1997:258-261.
17. Cush JJ, Kavanaugh A, Matteson EL. American College of Rheumatology.
Hotline. Available at:
www.rheumatology.org/publications/hotline/0305NSAIDs.asp. Accessed September
13, 2005.
18. Lander SA, Wallace DJ, Weisman MH. Celecoxib for systemic lupus
erythematosus: case series and literature review of the use of NSAIDs in SLE.
Lupus. 2002;11:340-347.
19. Toubi E, Rosner I, Rozenbaum M, et al. The benefit of combining
hydroxy-chloroquine with quinacrine in the treatment of SLE patients. Lupus.
2000;9:92-95.
20. Wallace DJ. Management of lupus erythematosus: recent insights. Curr Opin
Rheumatol. 2002;14:212-219.
21. Ting WW, Sontheimer RD. Local therapy for cutaneous and systemic lupus
erythematosus: practical and theoretical considerations. Lupus.
2001;10:171-184.
22. Gourley MF, Austin HA 3rd, Scott D, et al. Methylprednisolone and
cyclophosphamide, alone or in combination, in patients with lupus nephritis: a
randomized, controlled trial. Ann Intern Med. 1996;125:549-557.
23. Cunnane G, Lane NE. Steroid-induced osteoporosis in systemic lupus
erythematosus. Rheum Dis Clin North Am. 2000;26:311-329.
24. Reid IR. The roles of calcium and vitamin D in the prevention of
osteoporosis. Endocrinol Metab Clin North Am. 1998;27:389-398.
25. Boumpas DT, Austin HA 3rd, Vaughn EM, et al. Controlled trial of pulse
methylprednisolone versus two regimens of pulse cyclophosphamide in severe
lupus nephritis. Lancet. 1992;340:741-745.
26. Balow JE, Austin HA. Therapy of membranous nephropathy in systemic lupus
erythematosus. Semin Nephrol. 2003;23:386-391.
27. Nossent HC, Koldingsnes W. Long-term efficacy of azathioprine treatment
for proliferative lupus nephritis. Rheumatology. 2000;39:969-974.
28. Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in
patients with diffuse proliferative lupus nephritis. Hong Kong-Guangzhou
Nephrology Study Group. N Engl J Med. 2000;343:1156-1162.
29. Moroni G, Maccario M, Banfi G, et al. Treatment of membranous lupus
nephritis. Am J Kidney Dis. 1998;31:681-686.
30. Pisoni CN, Sanchez FJ, Karim Y, et al. Mycophenolate mofetil in systemic
lupus erythematosus: efficacy and tolerability in 86 patients. J Rheumatol.
2005;32:1047-1052.
31. Levy Y, Sherer Y, George J, et al. Intravenous immunoglobulin treatment of
lupus nephritis. Semin Arthritis Rheum. 2000;29:321-327.
32. Duna GF, Cash JM. Treatment of refractory cutaneous lupus erythematosus.
Rheum Dis Clin North Am. 1995;21:99-115.
33. Waknine Y. Orphan Drug Approvals: Bronchitol, Prestara, GTI-2040: Medscape
Medical News. 2005. Available at: www.medscape.com/viewarticle/509116.
Accessed September 15, 2005.
34. Shaver K. DHEA for lupus. Pharmacist's Letter. 2000;6:161210.
35. van Vollenhoven RF. Dehydroepiandrosterone in systemic lupus
erythematosus. Rheum Dis Clin North Am. 2000;26:349-362.
36. Anolik JH, Aringer M. New treatments for SLE: cell-depleting and
anti-cytokine therapies. Best Pract Res Clin Rheumatol. 2005;9:859-878.
37. Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel
treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial
of rituximab. Arthritis Rheum. 2004;50:2580-2589.
38. Van den Bergh B, Selleslag D, Boelaert JR, et al. Management of
therapy-resistant systemic lupus erythematosus with rituximab: report of a
case and review of the literature. Acta Clin Belg. 2005;60:102-105.
39. Lacy CF, Armstrong LL, Goldman MP. Drug Information Handbook. 12th ed.
Hudson, Ohio: Lexi-Comp; 2004:139-1021.
To comment on this article, contact
editor@uspharmacist.com.



