US Pharm. 2012;37(2):32-39.
Cardiovascular disease (CVD) remains the most common cause of death in the United States (25%). By 2008, about 27 million people in the U.S. were diagnosed with CVD, accounting for roughly one-half trillion dollars in health care expenditures.1,2 Atherosclerosis and thromboembolic disorders feature prominently in the pathogenesis of acute coronary syndromes (ACS), a spectrum of CVD that includes angina, myocardial infarction (MI), and stroke. Platelet activation plays a major role in normal homeostasis, but uncontrolled platelet activation leads to the formation of occlusive platelet-rich thrombi, which cause ischemic diseases such as MI, stroke, and peripheral arterial disease (PAD). There are many pathways that can activate platelets, and agents that can manipulate these pathways are the basis of pharmacotherapy for several thromboembolic conditions. This paper will review platelets’ involvement in the pathogenesis of thrombosis, available antiplatelet agents, and the pharmacist’s role in antiplatelet therapy.
Platelets’ Role in Homeostasis and Thrombosis
Platelets—blood cells formed in the bone marrow—figure significantly in the promotion of homeostasis. Under normal conditions, platelets circulate freely without adhering to each other or to the inner lining of undamaged blood-vessel endothelium. When injury occurs, damaging the endothelial lining, the subendothelial matrix and collagen are exposed. Subendothelial collagen is a strong thrombogenic substrate that provides a powerful stimulus for calcium release and induces procoagulant activity.3
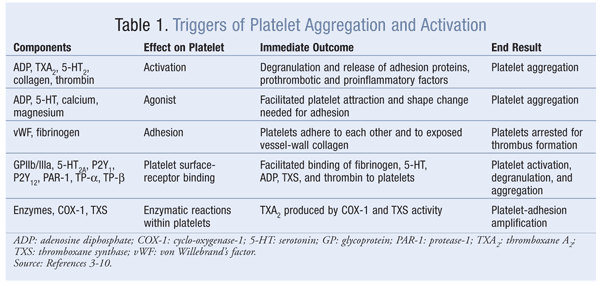
The initial phase of platelet adhesion at the injury site is mediated by the platelet’s integrin glycoprotein (GP) receptor complex (alpha-IIb/beta-III integrin, GPIIb/IIIa, and GPVI), which binds von Willebrand’s factor (vWF) and other ligands and mediates platelet adhesion to the exposed vessel-wall collagen. These interactions allow the platelet to be arrested, activated, and aggregated.3 Platelet stimulation resulting from adhesion further activates GPIIb/IIIa receptors, enabling the binding of soluble fibrinogen and vWF and the release of granules.3 Degranulation causes the granules to release serotonin (5-HT), epinephrine (a vasoconstrictor), adenosine diphosphate (ADP), and thromboxane A2 (TXA2).
Serotonin, a vasoconstrictor that binds 5-HT2A receptors on the platelet’s surface, amplifies the platelet’s response by stimulating it to change its shape and aiding recruitment to sites of injury.4 5-HT also promotes the retention of procoagulant proteins—fibrinogen and thrombospondin—on the platelet’s surface. 5-HT is involved in shear-induced aggregation and thrombus propagation through positive feedback from intraplatelet 5-HT stores.
ADP attracts additional platelets to the area via two ADP receptors—P2Y1 and P2Y12—on the platelet’s surface. The platelet’s change in shape and short-lived aggregation are mediated by P2Y1.5 Binding of ADP to the P2Y12 receptor causes signaling cascades, resulting in platelet aggregation and thrombus growth and stabilization.5 P2Y12 also is critical for the amplification of platelet aggregation induced by other agents, including 5-HT, TXA2, and thrombin.4 ADP contributes to platelet activation during protective homeostasis, at the initial platelet monolayer formation, and in pathologic thrombosis, when the occlusive platelet-rich thrombus is formed.
TXA2 is produced from arachidonic acid via enzymatic conversion by cyclo-oxygenase-1 (COX-1), and thromboxane synthase released from degranulating platelets binds to TXA2 receptors TP-alpha and TP-beta (platelet effects are mediated primarily through TP-alpha) to amplify adhesion response. This enables the change in platelet shape, recruitment enhancement, and aggregation of platelets to the primary platelet plug. The most important action of TXA2 released from activated platelets and accumulated at the site of atherosclerotic rupture is the contraction of microvessels downstream to interrupt blood flow.6 TXA2 activates platelets during protective hemostasis and pathologic thrombus formation.
Thrombin is a potent platelet agonist that activates platelets at extremely low concentrations (lower than that required for activation of the coagulation cascade).7,8 Thrombin-mediated platelet activation contributes to pathologic thrombosis through the formation of an occlusive platelet-rich thrombus.7,8 Thrombin activates platelets by binding protease-1 (PAR-1), the main thrombin receptor on human platelets, on the platelet’s surface.9 Platelet activation and stimulation of aggregation by PAR-1 are characterized by a high-affinity state and the release of ADP and TXA2, which leads to enhanced thrombus formation.10 This positive feedback continues and eventually results in a platelet plug, which combines with fibrin to form a platelet–fibrin clot. TABLE 1 lists triggers for platelet activation and aggregation.
The Platelet’s Role in Thrombotic Diseases
Platelets’ hyperactivity contributes to pathologic thrombus formation in several disease states.11 When atherosclerotic plaque ruptures, platelets adhere to the lipid core on the matrix, and exposed collagen is activated and then aggregates to form a prothrombotic surface that promotes thrombotic formation mediated by GP receptors, factors, and amplifiers. Thrombus formation in the microvascular system causes partial or complete arterial occlusion, and the resultant obstruction of blood flow is the hallmark of several CVDs, such as unstable angina, MI, stroke, and PAD. Typically, platelet-rich white thrombi are not fully occluded, and they are associated with unstable angina, non-ST segment elevation (NSTE) ACS, or NSTE MI (NSTEMI). Progression to a completely occluded thrombus mediated by the coagulation cascade involves formation of a fibrin-rich red clot superimposed on the underlying platelet-rich white thrombus; this is usually found in STE ACS or STE MI (STEMI).
Systemic inflammatory syndrome (SIS) is related to sepsis and thrombotic diseases such as stroke, PAD, and diabetes.12 SIS causes disseminated intravascular platelet activation with inflammatory-response amplification through the release of inflammatory cytokines and growth factors, resulting in microvascular thrombosis.13 In diabetes mellitus, which is characterized by hyperglycemia and insulin resistance, the systemic and cellular environments are modified to generate a prothrombotic state.14 This dysfunctional metabolic environment promotes the development of the atherosclerotic process, which involves the coagulation cascade, endothelial-lining dysfunction, and platelet adhesiveness, and contributes to pathologic thrombus formation.
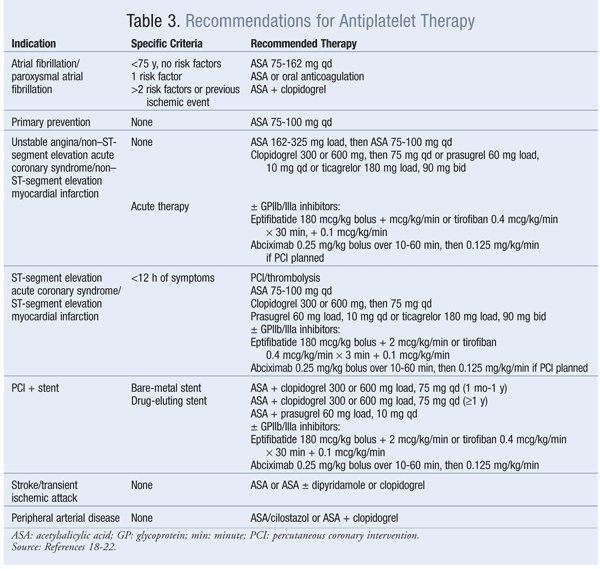
Given the striking role of platelet activation and its endothelial interaction in the pathogenesis of atherothrombotic conditions, antiplatelet therapy is beneficial in both primary and secondary prevention of atherothrombotic conditions associated with many CVDs.13 The possibility of reversible or irreversible modulation of platelet activation has led to modalities that are continuously being exploited in the design of antiplatelet therapy. Antiplatelet therapy is recommended in several clinical-practice guidelines for the prevention and treatment of stroke, NSTEMI, STEMI, unstable angina, PAD, and diabetes.
Available Agents
Antiplatelet drugs utilize a variety of mechanisms to interfere with platelet activation and aggregation.15 Mechanisms include the targeting and inhibition of key pathways of platelet activation and aggregation (including inhibition of various platelet-surface receptors, such as COX-1, P2Y12, and GPIIb/IIIa). Another pathway that inhibits platelet activation and aggregation is greater adenylate cyclase activity, with a corresponding increase in cyclic adenosine monophosphate (cAMP) and guanosine monophosphate levels and a reduction in phosphodiesterase (PDE).
Thromboxane Inhibitors: Aspirin irreversibly inhibits COX-1, thereby reducing the production of TXA2 in platelets.16 TXA2 is a powerful inducer of platelet aggregation. This inhibition occurs in aspirin doses as low as 30 mg, but doses of 75 mg to 325 mg are effective for preventing thrombotic events. Aspirin resistance may occur in up to 40% of patients, resulting in an increased risk of CV events. Aspirin resistance may be triggered by inadequate dose, drug interactions, genetic variations in COX-1 enzyme and TXA2 activity, and increased platelet turnover.16
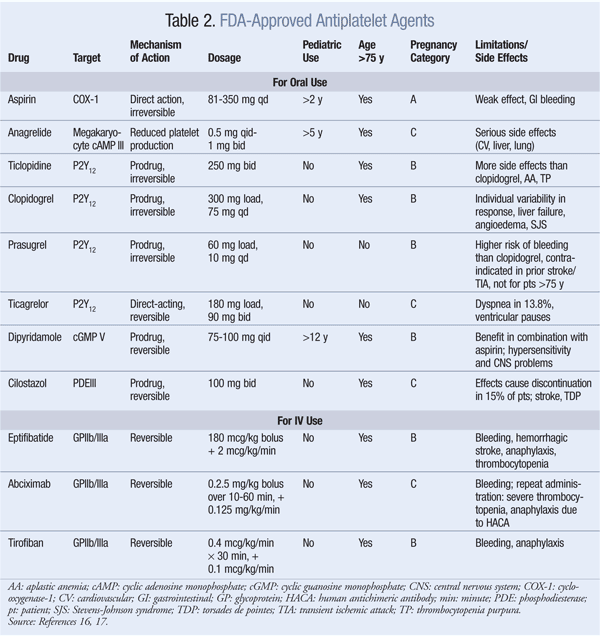
ADP Receptor Antagonists: ADP receptor antagonists inhibit the P2Y12 receptor on the platelet’s surface, thus preventing fibrinogen from binding to platelets, a necessary step in aggregation and thrombus formation.16 There are two subgroups of agents: thienopyridines—which include ticlopidine, clopidogrel, and prasugrel—and ticagrelor, a nonthienopyridine agent. These agents are eliminated renally, except for ticagrelor, which is eliminated via the feces. Unlike the others, ticagrelor is not a prodrug, and it has the shortest duration of antiplatelet effects. Prasugrel and ticagrelor have no significant drug interactions compared with ticlopidine and clopidogrel.
GPIIa/IIIb Receptor Inhibitors: Eptifibatide, abciximab, and tirofiban reversibly inhibit platelet aggregation by preventing the binding of vWF and fibrinogen to GPIIb/IIIa receptors on the platelet’s surface.16 These agents are given IV in conjunction with other anticoagulants, thus contributing to the increased risk of bleeding. Other side effects include thrombocytopenia and anaphylaxis in some patients taking abciximab.
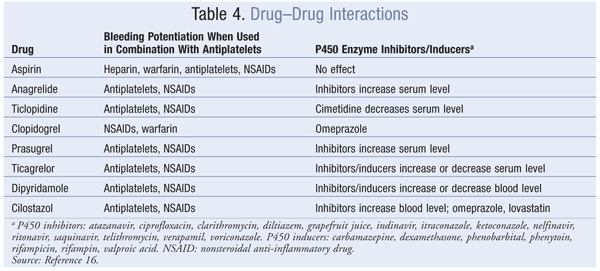
cAMP: Anagrelide and cilostazol inhibit cAMP PDEIII receptors, thereby suppressing platelet aggregation.16 Anagrelide, which is indicated for thrombocythemia due to myeloproliferative disease, is contraindicated in congestive heart failure. Serious side effects include CV, pulmonary, and central nervous system problems. Cilostazol is indicated for PAD, and CV side effects are prevalent. Dipyridamole, which stimulates cAMP levels and inhibits platelet aggregation, is indicated as an adjunct to anticoagulation to prevent postoperative thrombosis associated with heart valve replacement. A major concern with dipyridamole is hypersensitivity reactions. Antiplatelet agents, uses, monitoring parameters, and drug–drug interactions are listed in TABLES 2, 3, and 4, respectively.
The Pharmacist’s Role
Pharmacists must take a greater role in the management of patients receiving or requiring antiplatelet therapy. Pharmacists’ knowledge of medications and their clinical, communication, and patient-care skills will allow them to fulfill this role, including making sure that the progression from inpatient care (where antiplatelet drugs are prescribed) to outpatient care is seamless and optimized. This is crucial since 17% of patients requiring antiplatelet therapy will delay filling their prescription, another 1% will abandon their filled antiplatelet medication, and 79% of patients with risk factors for thromboembolic disease or CVD do not receive any form of antiplatelet therapy.23,24 Pharmacists are important in the continuum of therapy and in the patient’s transition from the inpatient setting to outpatient care.
As a member of the health care team, the pharmacist should ensure that the appropriate agent is selected for the patient in accordance with current guidelines. During the transition from inpatient to outpatient care, the pharmacist should provide the patient and family members or caregiver with the medication discharge plan, including the antiplatelet prescription and information about dosing and frequency, therapy duration, and side effects.
In cooperation with the patient, the pharmacist should select the pharmacy where the medication will be filled to ensure that the patient receives the medication and takes it home ready for administration. The pharmacist should follow up with the patient within a week to answer any questions the patient may have about indication, dosing and frequency, and side effects. The pharmacist should also help eliminate delays in filling prescriptions in the outpatient setting, ensure that prescriptions are picked and available for administration, and prevent premature discontinuation of the medication. Pharmacists can provide community medication therapy management (MTM) services during which the patient and family members or caregiver are re-educated about the medication and regimen, how to manage compliance, drug–drug and drug–food interactions, and side effects. Patient monitoring to prevent complications is critical, and the pharmacist, in conjunction with other providers, should assess the efficacy and safety of therapy by direct measurement of platelet activity, if possible; otherwise, total blood counts (including platelets and measurement of hepatic and renal function) should be performed. Other monitoring parameters include reviewing symptoms, administering questionnaires, and asking about the patient’s health and any need for changes or referral back to the medical provider. Constant communication between providers is essential for the provision of continuum-of-care services.
Conclusion
Platelet aggregation plays an important role in thromboembolic disorders, and antiplatelet therapies have been shown to reduce the morbidity and mortality associated with these disorders. Antiplatelet agents are recommended for clinical management of thromboembolic disorders for both primary and secondary prevention of further thrombosis.
Pharmacists are in a unique position to provide a continuum of care and improve desired outcomes for patients receiving antiplatelet therapy. This can be accomplished with MTM services, which should include patient education and continued contact with the patient, monitoring for bleeding and other interactions, and communication of findings to the patient’s care team to resolve and/or avoid adverse therapy outcomes.
REFERENCES
1. CDC. Heart disease. www.cdc.gov/heartdisease. Accessed October 12, 2011.
2. CDC. Heart disease and stroke prevention. www.cdc.gov/chronicdisease/
3. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99:1293-1304.
4. Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403-412.
5. Dorsam RT, Kunapuli SP. Central role of the P2Y12 receptor in platelet activation. J Clin Invest. 2004;113:340-345.
6. Goto S. Propagation of arterial thrombi: local and remote contributory factors. Arterioscler Thromb Vasc Biol. 2004;24:2207-2208.
7. Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100:148-152.
8. Mann KG. Thrombin formation. Chest. 2003;124:4-10S.
9. Rauch U, Osende JI, Fuster V, et al. Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences. Ann Intern Med. 2001;134:224-238.
10. Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070-1077.
11. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482-2494.
12. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378-3384.
13. Löwenberg EC, Meijers JC, Levi M. Platelet-vessel wall interaction in health and disease Neth J Med. 2010;68:242-251.
14. Mafrici A, Proietti R. Atherothrombosis in patients with type 2 diabetes mellitus: an overview of pathophysiology. G Ital Cardiol (Rome). 2010;11:467-477.
15. Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74:597-607.
16. Micromedex Solutions online database [subscription required]. Healthcare Thompson Reuters 2012.
17. Drugs@FDA. www.accessdata.fda.gov/
18. Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (updating the 2006 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;123:104-123.
19. Wright RS, Anderson JL, Adams CD, et al. 2011 ACCF/AHA focused update of the guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;123:2022-2060.
20. Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227-276.
21. Rooke TW, Hirsh AT, Misra S, et al. 2011 ACCF/AHA focused update of the guideline for the management of patients with peripheral artery disease (updating the 2005 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;58:2020-2045.
22. Kushner FG, Hand M, Smith SC Jr, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2009;120:2271-2306.
23. Ho PM, Tsai TT, Maddox TM, et al. Delays in filling clopidogrel prescription after hospital discharge and adverse outcomes after drug-eluting stent implantation: implication for transitions of care. Circ Cardiovasc Qual Outcomes. 2010;3:261-266.
24. Shrank WH, Choudhry NK, Fischer MA, et al. The epidemiology of prescriptions abandoned at the pharmacy. Ann Intern Med. 2010;153:633-640.
To comment on this article, contact rdavidson@uspharmacist.com.


