US Pharm. 2010;35(6):25-25.
Cushing’s syndrome (CS) is a manifestation of symptoms that are precipitated by excessive cortisol production. CS is a rare but serious condition that can be fatal. There are several known causes of the syndrome, and treatments include surgery, radiation, and drug therapy. Diagnosing the specific origin of CS is complex and can be expensive, but diagnostic tests are essential in order to effectively manage a patient’s therapy.
EPIDEMIOLOGY
This syndrome is named for Harvey Williams Cushing, an American neurosurgeon who became known in 1912 for his work on the pituitary gland.1 The incidence of new cases of CS in the United States ranges from 0.7 to 2.4 cases per million people per year. This translates to an estimated 200 to 700 new cases annually.2 Drug-induced CS is believed to be the most prominent form of this syndrome, and there are numerous documented cases.3,4 Recent studies in which type 2 diabetic patients and obese patients were screened suggest that CS may be more common than previously thought.3 In patients with hypertension or poorly controlled blood glucose, CS rates as high as 2% to 5% have been documented.3,5,6
ETIOLOGY
Corticotropin-releasing hormone (CRH), produced by the hypothalamus, stimulates the production and release of corticotropin (ACTH) through its actions on the anterior pituitary. ACTH then acts on the adrenal cortex to release cortisol (FIGURE 1).7 Cortisol regulates the breakdown of fat and the metabolism of proteins and carbohydrates. Cortisol production increases prior to waking and peaks within a few hours after waking. Cortisol levels are lowest before sleep and during the first few hours of sleep.8
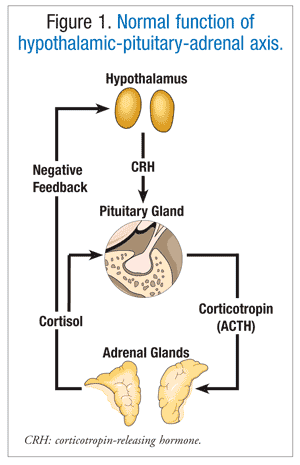
Hypercorticism refers to an excess of corticosteroids in the body. This excess of corticosteroids—specifically, cortisol—can lead to CS. The excess cortisol is rarely due to spontaneous cortisol production by the adrenal gland.8 Typical causes include tumors and drugs. A patient who has CS may present with one or more of the following: moon face, central obesity (trunk accumulation of adipose tissue), myopathy, muscle weakness, fatigue, hirsutism, osteoporosis, amenorrhea, hypertension, psychiatric symptoms, striae (purple stripes or lines in skin), back pain, and/or diabetes mellitus.3,9 CS can have ACTH-dependent or ACTH-independent origins.3
ACTH-Dependent CS
Excessive cortisol release due to increased ACTH levels is defined as ACTH-dependent CS. A major cause of ACTH-dependent CS is the presence of ACTH-dependent tumors normally found in the pituitary gland.3 Pituitary adenomas, which account for approximately 70% of dependent causes, are typically small and benign.3 Ectopic tumors are the other major cause of ACTH-dependent CS. These ectopic tumors occur in abnormal places (i.e., lung, kidney) and account for about 15% of ACTH-dependent CS cases.3 Ectopic tumors outside the hypothalamus constitute less than 1% of ACTH-dependent cases. These nonhypothalamic tumors excrete CRH, which leads to the overproduction of ACTH.3
ACTH-Independent CS
Cases of ACTH-independent CS present with increased cortisol levels that are not related to increased ACTH levels. ACTH-independent CS is further broken down by cause—either exogenous or endogenous.
Exogenous CS: Drug-induced, or exogenous, CS occurs when an excess of steroids is introduced into the body. Drug-induced CS is the most common cause of the syndrome.3
Endogenous CS: There are several types of ACTH-independent tumors. Adrenal adenomas and carcinomas are the most common independent tumors. Other independent causes, occurring in less than 1% of cases reported, include bilateral macronodular hyperplasia and primary pigmented nodular adrenocortical disease.3
DIAGNOSIS
To diagnose CS, a history is obtained and a physical examination is conducted to assess the presenting signs and symptoms. There are currently two first-line screening tests for CS, with a third screening test currently being validated. The first-line screening tools are the 24-hour urinary free cortisol (UFC) test and the low-dose dexamethasone suppression test (DST).7
Confirmation of CS
UFC Test: UFC levels correlate with free plasma levels of cortisol. The patient collects three 24-hour samples of urine for analysis.10 Normal urine cortisol tests are under 100 mcg/24 h.11 A result that is four times the upper limit of normal (typically 440 mcg/24 h), which is rare, indicates CS.10 The UFC test is not indicated in patients with renal impairment—defined as a glomerular filtration rate less than 30 mL/min—because of decreased cortisol excretion. The screening sensitivity does not allow for the detection of subclinical or preclinical CS.
DST: In healthy patients, dexamethasone suppresses plasma cortisol levels. In CS, this mechanism can be overridden by the underlying disease. For the DST, the patient is administered 1 mg dexamethasone between 2300 and 2400 hours. The following morning, cortisol concentration levels are measured. A measurement above 5 mcg/dL indicates inhibition of a negative feedback loop, as seen in CS. While a level of 1.8 mcg/dL allows for identification of pseudo-CS, cyclic CS, or early stages of CS, it also may create false-positive results.12 A two-day 2-mg DST may be used in clinical practice; however, cost and increased hospitalization are limiting factors in its use.
Late-Night Salivary Cortisol Test (LST): LST is a new screening tool being evaluated for CS. Salivary cortisol concentration is closely related to free plasma, but it remains independent of salivary flow rate. Normal ranges for this screening are assay-dependent. Current studies suggest high specificity and sensitivity, but further research is needed to validate the use of LST as a screening tool.10
Differential Diagnosis
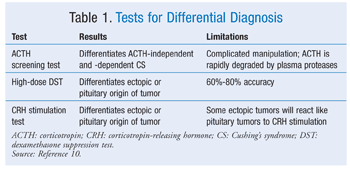
Once CS is diagnosed, further testing is necessary to differentiate the cause, thereby enabling proper treatment. Current second-line tests are ACTH measurement, high-dose DST, and the CRH stimulation test (TABLE 1).10 All patients with ACTH-dependent CS should undergo pituitary MRI. The bilateral inferior petrosal sinus sampling (BIPSS) test samples blood surrounding the pituitary, allowing for accurate diagnosis of pituitary origin of high ACTH. BIPSS also is recommended in patients with ACTH-dependent CS. However, owing to rare but significant complications, the risks and benefits need to be taken into consideration. BIPSS has a sensitivity of 95% to 99% when performed at “experienced centers.”10 Several new differential diagnosis tests are currently being researched, including one involving the use of desmopressin.10
Complications of CS
Accurate treatment of CS is important in order to avoid the numerous complications that can stem from disease progression. Major cardiovascular complications are hypertension, impaired glucose tolerance, obesity, hyperlipidemia, hypercoagulability, metabolic syndrome, and osteoporosis. Psychiatric complications such as psychosis, mania, suicidal tendency, obsessive-compulsive disorder, cognition changes, and anxiety also may develop. Last, high cortisol levels may create alterations in other areas of the endocrine system, such as the somatotrophic axis (which can decrease growth rate in younger patients or alter fat distribution in adults), the gonadal axis (which can have adverse effects on reproductive organs), and the thyroid axis (which can lead to hypothyroidism or hyperthyroidism). These complications may persist even if a patient has subclinical CS.10
TREATMENT
Drug-induced CS typically is treated by discontinuing the causative medication. Problems arise in complex patients (e.g., transplant patients), in whom discontinuation of the steroid medication may lead to serious adverse outcomes. Surgery, radiation, and pharmacotherapy constitute the standard treatment for tumor-related causes of CS. Following treatment, further screenings are necessary to assess the success of therapy.2,13
Nonpharmacologic
The first-line option for pituitary tumors is transsphenoidal surgery (technique in which the pituitary tumor is removed through the nose, leaving remaining pituitary function intact), which gives the patient a chance of remission of CS. Surgical remission rates range from 60% to 80%, but this number may vary owing to different definitions of remission.2 The average recurrence rate is around 20%.2 As with surgical remission, there is no current collaborated definition of a cure for CS.2 Surgery or radiation with or without adjunctive pharmacologic treatment is currently seen in clinical practice.
Pharmacologic
Current pharmacologic treatment does not correct the underlying cause of excessive cortisol, which typically limits its use to adjunctive therapy. However, pharmacologic monotherapy may be necessary when surgery or radiation is not appropriate or is delayed. Three pharmacologic strategies exist to treat CS.
Inhibition of Steroid Synthesis: The first strategy is the inhibition of steroid synthesis. Drugs used in this strategy include ketoconazole, mitotane, metyrapone, and etomidate.13 These drugs, through different mechanisms, block the conversion of cholesterol to cortisol. This causes a decrease in cortisol levels and the resolution of CS symptoms. There are several disadvantages to this strategy: Higher-than-normal doses of medications are used, patients rarely remain in remission after discontinuing the medication, and steroid replacement therapy may be needed to avoid adrenal insufficiency.14 None of these agents is FDA-approved to treat CS, and the doses reported in the literature cover a wider-than-normal range, indicating the need for very careful attention to side effects.
Ketoconazole. A common side effect of ketoconazole, an imidazole antifungal agent, is inhibition of the CYP450 pathway.15 This inhibition halts the production of cortisol, leading to a reversal of CS symptoms. Ketoconazole therapy is initiated at a daily dose of 400 mg to 600 mg. The dose is titrated until a level of 1,200 mg to 1,600 mg, divided into four equal doses, is reached.16,17 Ketoconazole’s safety and efficacy support its use as a first-line choice for many prescribers.16,17
Mitotane. An antineoplastic agent, mitotane (Lysodren) inhibits cortisol synthesis by inhibiting 18-hydroxylase and 3-beta-hydroxysteroid dehydrogenase. At high doses, mitotane has adrenolytic activity (breaks down adrenaline), which causes adrenal cortical atrophy. Mitotane is started at 0.5 g to 1 g daily and increased every 1 to 4 weeks.13 At doses greater than 4 g daily, the adrenal cortex may be irreversibly impaired. Mitotane’s half-life is 18 to 159 days, and the drug has been detected in adipose tissue as long as 22 months after treatment.
Metyrapone. Metyrapone (Metopirone), a diagnostic agent, works by blocking 11-beta-hydroxylase, halting cortisol production. Metyrapone can be used as monotherapy or as adjunctive therapy with radiation. Typical dosing of metyrapone begins at 0.5 g to 1 g daily, increasing every few days to a maximum dose of 6 g.13 Blocking 11-beta-hydroxylase leads to increased ACTH, resulting in increased androgenic and mineralocorticosteroid precursors. This can lead to adverse effects such as hypertension, acne, hirsutism, and nausea. Although this agent is not yet available in the U.S., compassionate use is an option.16
Etomidate. Etomidate (Amidate), an imidazole-derivative anesthetic, is used for the induction and maintenance of anesthesia. It blocks cholesterol’s side-chain cleavage and 11-beta-hydroxylase, inhibiting the conversion of cholesterol to cortisol. Etomidate is the only medication in this strategy that can be given IV, and case reports have cited a significant reduction in cortisol within 12 hours.18 Because of its anesthetic effects, careful dose titration is required. An effective dosage range has been reported to be 0.01 mg/kg/h to 0.1 mg/kg/h.18
Modulation of Pituitary ACTH: The second strategy is to modulate pituitary ACTH release, causing a decrease in cortisol secretion from the adrenal gland. This strategy has a poor response rate, and no large-scale, placebo-controlled studies have been reported. As with the drugs used to inhibit steroid synthesis, none of these agents is FDA-approved for the treatment of CS. Drugs used include cyproheptadine, octreotide, and valproic acid.
Inhibition of Cortisol’s Action: The third strategy involves using an agent that blocks cortisol’s action. Mifepristone competitively inhibits glucocorticoid, androgen, and progestin receptor binding. No large clinical trials have been performed to assess the benefit of this drug, but a few investigational studies of ectopic CS have shown a potential benefit.19,20 However, dosage adjustment was found to be difficult. Currently, mifepristone is indicated only as an abortifacient in the U.S. Its use in CS is an off-label indication, and further testing is warranted.
PHARMACIST INVOLVEMENT
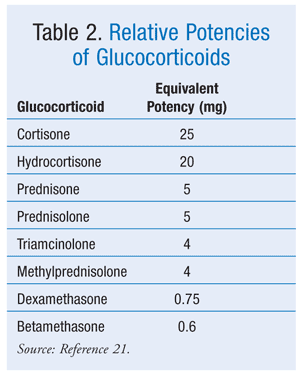
CS treatment is not often seen in community pharmacy practice. Cases of CS are rare, and often they can be corrected with simple dosing changes. Chronic steroid therapy (e.g., for asthma, organ transplantation) and inappropriate drug use are situations seen in an outpatient setting. Proper steroid conversions and patient education are critical roles for the pharmacist to perform. A steroid conversion chart (TABLE 2) is a helpful guide for regimen changes.21 Patients should also be educated about their dosing schedule and the appropriate treatment duration.8
CONCLUSION
CS is a rare condition with potentially serious complications if it is left untreated. Proper diagnosis is important to assess the underlying cause of CS, allowing for appropriate treatment. Pharmacologic treatment, while not first-line, plays a vital role for selected patients with CS. Pharmacist involvement in the management of patients’ steroid therapy can lead to fewer cases of drug-induced CS.
REFERENCES
1. Merriam-Webster MedlinePlus Medical Dictionary. Cushing’s disease. www.merriam-webster.com/medlineplus/cushings%20disease. Accessed October 6, 2009.
2. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605-1617.
3. UpToDate. Overview of the treatment of Cushing’s syndrome. http://newadonis.creighton.edu/hsl/uptodate/~gmD018.asp [subscription required]. Accessed October 8, 2009.
4. Stevens DJ. Cushing’s syndrome due to the abuse of betamethasone nasal drops. J Laryngol Otol. 1988;102:219-221.
5. Leibowitz G, Tsur A, Chayen SD, et al. Pre-clinical Cushing’s syndrome: an unexpected frequent cause of poor glycaemic control in obese diabetic patients. Clin Endocrinol (Oxf). 1996;44:717-722.
6. Catargi B, Rigalleau V, Poussin A, et al. Occult Cushing’s syndrome in type-2 diabetes. J Clin Endocrinol Metab. 2003;88:5808-5813.
7. Orth DN. Cushing’s syndrome. N Engl J Med. 1995;332:791-803.
8. Fitzgerald PA. Endocrine disorders. In: McPhee SJ, Papadakis MA, Tierney LM Jr, eds. CURRENT Medical Diagnosis & Treatment. www.accessmedicine.com.cuhsl.creighton.edu/content.aspx?aID=14198 [subscription required]. Accessed October 6, 2009.
9. Kelly W. Cushing’s syndrome: a review of diagnosis and treatments. Sudan Med J. 2009;45:3-7.
10. Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2003;88:5593-5602.
11. Pagana K, Pagana T. Mosby’s Diagnostic and Laboratory Reference. 9th ed. St. Louis, MO: Mosby; 2009:303.
12. Giraldi FP, Ambrogio AG, De Martin M, et al. Specificity of first-line tests for the diagnosis of Cushing’s syndrome. Assessment in a large series. J Clin Endocrinol Metab. 2007;92:4123-4129.
13. Nieman LK. Medical therapy of Cushing’s disease. Pituitary. 2002;5:77-82.
14. Orth DN. Metyrapone is useful only as adjunctive therapy in Cushing’s disease [editorial]. Ann Intern Med. 1978;89:128-129.
15. Loose DS, Kan PB, Hirst MA, et al. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983;71:1495-1499.
16. Biller BMK, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93:2454-2462.
17. Winquist EW, Laskey J, Crump M, et al. Ketoconazole in the management of paraneoplastic Cushing’s syndrome secondary to extopic adrenocorticotropin production. J Clin Oncol.
18. Schulte HM, Benker G, Reinwein D, et al. Infusion of low dose etomidate: correction of hypercortisolemia in patients with Cushing’s syndrome and dose-response relationship in normal subjects. J Clin Endocrinol Metab. 1990;70:1426-1430.
19. Nieman LK, Chrousos GP, Kellner C, et al. Successful treatment of Cushing’s syndrome with the glucocorticoid antagonist RU 486. J Clin Endocrinol Metab. 1985;61:536-540.
20. Chu JW, Matthias DF, Belanoff J, et al. Successful long-term treatment of refractory Cushing’s disease with high-dose mifepristone (RU 486). J Clin Endocrinol Metab. 2001;86:3568-3573.
21. Gums JG, Anderson S. Adrenal gland disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 7th ed. New York, NY: McGraw-Hill; 2008:1265-1280.
To comment on this article, contact rdavidson@uspharmacist.com.



