US Pharm. 2012;37(1)(Oncology suppl):2-7.
ABSTRACT: Life-threatening and dose-limiting toxicities commonly associated with chemotherapy have led to an increased interest in the role of pharmacogenomics in developing individualized treatments for patients based on their genetic biomarker profile for toxicities. Genetic polymorphisms associated with drug-metabolizing enzymes, drug transporters, or drug targets affect the pharmacokinetics and/or the pharmacodynamics of many oncology regimens. The early integration of pharmacogenomics in chemotherapy offers clinicians the ability to better predict and manage drug toxicities, improve patients’ quality of life, and maximize the efficacy of treatment.
One of the biggest challenges in chemotherapy is managing the toxicities associated with treatment. Many severe toxicities, such as myelosuppression and renal, hepatic, cardiovascular, gastrointestinal (GI), pulmonary, and central nervous system (CNS) effects, can be dose limiting and even life threatening, and often severely compromise a patient’s qualify of life and frequently jeopardize well-planned therapeutic regimens. A drug’s efficacy and the manifestation of its toxicities vary significantly among individual patients undergoing chemotherapy.1 For example, clinical evidence indicates that the steady-state levels of 6-mercaptopurine (6-MP) in acute lymphocytic leukemia (ALL) patients can range up to 10-fold or higher among cancer patients with the same administered drug dose.2
When designing and selecting a cancer therapeutic regimen, the acute and cumulative long-term, life-threatening, and dose-limiting toxicities associated with cancer therapy must be considered. Factors that can affect drug efficacy and toxicities that should be taken into consideration are summarized in FIGURE 1; they include morphometric, demographic, pharmacologic, pharmacogenomic (PGx), physiological, and pathophysiological factors. In particular, numerous clinical studies indicate that interracial and interindividual polymorphisms in genes encoding for drug-metabolizing enzymes (DMEs), drug transporters, and drug targets are linked to the toxic clinical presentation of cancer patients.3
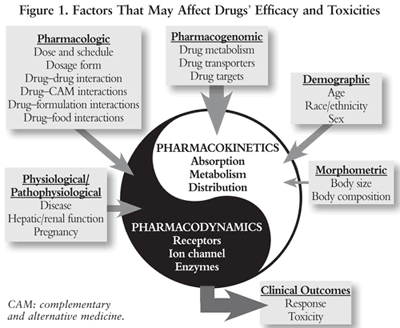
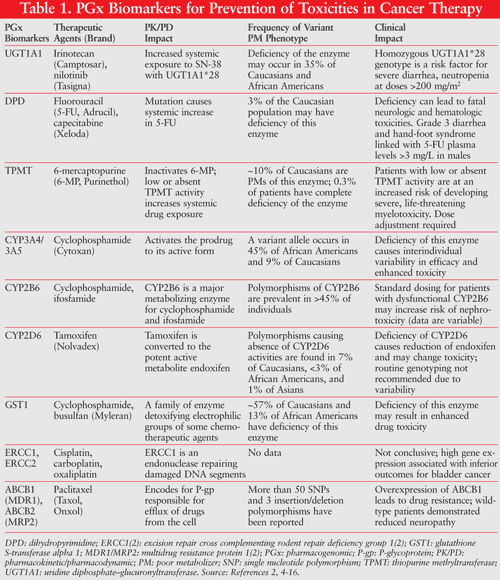
Genetic polymorphisms define the difference in the DNA sequence that occurs in more than 1% of the population. Over 2 million single nucleotide polymorphisms (SNPs; genetic polymorphism between two genomes that is based on a deletion, insertion, or exchange of a single nucleotide) have been identified in the human genome. Although a majority of the SNPs have no clinical significance, many of them contribute to clinically significant changes in drug pharmacokinetics and/or pharmacodynamics of cancer treatment. Genetic polymorphisms in genes that encode drug-metabolizing enzymes, drug transporters, drug targets, and any other molecules that affect drug disposition have been linked to interindividual differences in the toxicity of many chemotherapy agents. TABLE 1 lists some key biomarkers associated with potentially severe cancer treatment toxicities, many of which are cited in FDA-approved drug labels.2,4-16 Common biomarkers include gene variants, functional deficiencies, expression changes, and drug suppression. Most toxicity biomarkers can be categorized as either selective biomarkers for preventing toxicities or biomarkers for drug interactions that increase toxicity of the chemotherapeutic drugs.4
Most of the biomarkers for drug toxicity are DMEs. Based on their ability to metabolize, genetic polymorphisms in DMEs can be grouped into four categories: 1) extensive metabolizers (EMs), defined as individuals with normal and efficient drug metabolism; 2) poor metabolizers (PMs), defined as individuals with deficiencies in drug metabolism, which are typically caused by mutation or deletion of both alleles of a wide-type gene; 3) intermediate metabolizers (IMs); and 4) ultrarapid metabolizers (UMs), represented by a wide range in the levels of a specific enzyme activity from adequate to overexpression due to gene amplification. Standard doses of drugs with a steep dose-response relationship or a narrow therapeutic index (NTI) may produce adverse drug reactions, toxicity, or decreased efficacy in PMs. In contrast, when taken by UMs, that standard dose may be inadequate to produce the desired therapeutic effect.17
The most important example of a biomarker associated with cancer therapy is thiopurine methyltransferase (TPMT), which is a metabolizing enzyme responsible for 6-MP inactivation. Approximately 10% of Caucasians have partial deficiency in TPMT enzyme activity, while 0.3% of the patients have a complete deficiency in the enzyme activity. A PM of TPMT is at significant risk for severe drug toxicity associated with 6-MP.18 The FDA recommends a dosage reduction for patients with heterozygous or homozygous mutations in TPMT.
PGx Biomarkers for Toxicities Associated With Cancer Therapy
PGx research gives clinical practitioners the ability to predict potential toxicities and to improve therapeutic outcomes by utilizing the patient’s genetic profile of biomarkers to identify which patients are at greater risk for an adverse therapeutic response to a specific drug. By identifying the biomarkers associated with severe and potentially life-threatening drug toxicity for cancer patients, PGx therapy offers clinicians the potential to individualize and optimize treatment regimens in order to minimize toxicity and maximize drug response.19
It is important to identify those individuals with genetic biomarkers that indicate high risk for drug toxicity and low response rate from standard doses of cancer treatment regimens (TABLE 1). These biomarkers can be divided into three major categories: 1) DMEs, 2) drug transporters, and 3) drug targets based on their biological functions. Biomarkers are usually used in the appropriate therapy selection associated with targeted therapeutic agents directed at tumor cells with particular protein characteristics that differ significantly from those of their normal cell counterparts. PGx biomarkers can also be used by physicians to predict the severity of toxicity and to monitor the potential side effects of a patient’s treatment, based on his or her genetic profile. Thus, cancer therapy guided by PGx information has the potential to be more selective for cancer cells than normal cells. This PGx-based monitoring can also significantly improve the prognosis of cancer patients and potentially decrease the toxic effects of anticancer drugs on normal cells.4,19
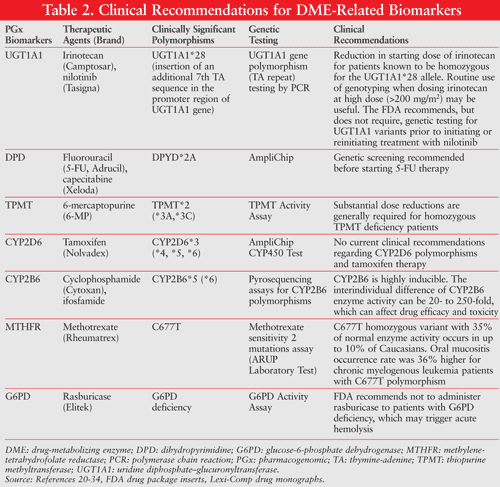
DME-Related Biomarkers
DMEs play an important role in inactivating many chemo-therapeutic drugs and in activating some chemotherapeutic prodrugs. The genetic polymorphisms of these drug metabolism–related potential biomarkers exert important effects on drug efficacy and sensitivity to toxicity. TABLE 2 outlines the clinical implication of polymorphism in chemotherapeutic agent–metabolizing enzymes, such as CYP450, glutathione S-transferase (GST), uridine diphosphate (UDP)–glucuronosyltransferase (UGT), TPMT, and dihydropyrimidine dehydrogenase (DPD).4,19-34
6-MP and TPMT Polymorphisms: 6-MP is used extensively in maintenance chemotherapy for childhood ALL. The rate and extent of the conversion of 6-MP to its active form, 6-thioguanine, can have a significant impact on 6-MP efficacy and toxicity. 6-thioguanine can be incorporated into DNA and ultimately inhibit cell replication. 6-MP can also be converted to 6-methylmercaptopurine via an S-methylation reaction catalyzed by TPMT. The extent and magnitude of this reaction can lead to significant changes in the levels of active metabolite and can cause substantial drug-related toxicities. Most large-scale PGx studies indicate that TPMT activity is highly variable and polymorphic, with approximately 90% of individuals having high activity (EMs), 10% with intermediate activity (IMs), and 0.3% with low or complete deficiency of the enzyme activity (PMs). Although this polymorphism is relatively rare, deficiency in TPMT leads to excessive levels of 6-thioguanine with consequently severe, potentially life-threatening hepatic toxicity, GI toxicity, and myelosuppression when exposed to standard doses of 6-MP. Deficiency of TPMT also increases the risk of secondary cancers associated with 6-MP standard-dose treatment.2,4,9,26,27
Clinical evidence indicates that there can be as much as a 10-fold variation in the steady-state dosage of 6-MP between TPMT PMs and EMs. Thus, the consequence of using standard dosage for patients with TPMT deficiency could be fatal. A TPMT genetic test has been initiated in the effective clinical management of patients with ALL in some medical centers (TABLE 2). Clinical evidence indicates that decreasing the dose of 6-MP by 10- to 15-fold in TPMT PM patients compared with conventional doses enables successful treatment without substantial toxicity.2,4,9,26,27
DPD and 5-Fluorouracil: 5-Fluorouracil (5-FU) is an injectable antimetabolite pyrimidine analogue used in the treatment of colon, rectal, breast, ovarian, and GI cancers (e.g., anal, esophageal, pancreatic, gastric) and head and neck cancers. The enzyme responsible for the catabolism of 5-FU is DPD, which is the rate-limiting step involved in the nucleotide metabolism of the pyrimidines uracil and thymine. DPD deficiency is an autosomal recessive metabolic disorder in which the dihydropyrimidine dehydrogenase enzyme activity is absent or significantly diminished in cells. Individuals with the DPD condition may develop life-threatening toxicity following exposure to 5-FU or the widely prescribed oral prodrug fluoro-pyrimidine capecitabine.7,8,13,24,25
More than 30 SNPs have been identified within the DPD gene. The most common variant that has been reported in approximately 45% of people with partial or complete DPD deficiencies is DPYD*2A.13 Complete DPD deficiency is relatively uncommon, occurring in less than 5% of the Caucasian population. African Americans have a markedly reduced mean DPD enzyme activity and a higher partial DPD deficiency when compared with Caucasians. Screening for DPYD*2A is commercially available and accounts for approximately 45% of all DPD deficiencies (TABLE 2). In addition, patients with partial or complete DPD activity can be identified by testing of their blood lymphocytes, which will also identify patients potentially at risk for 5-FU–related toxicity.7,8,13,24,25
UGT1A1 and Irinotecan: Irinotecan is a topoisomerase I inhibitor approved by the FDA for the treatment of advanced colorectal cancer and various other solid tumors. Irinotecan is a prodrug that requires activation by carboxylesterase to its active metabolite, SN-38. Furthermore, SN-38 is converted to inactive metabolite SN-38G by glucuronidation via the 1A1 isoform of UDP-glucuronosyltransferase (UGT1A1). Dose-limiting toxicities of irinotecan are diarrhea and neutropenia. These toxicities, which are associated with increased levels of SN-38, can be caused by a UGT1A1*28 polymorphism that may occur in 35% of Caucasians and African Americans. The UGT1A1*28 allele is associated with reduced UGT1A1 gene expression, and leads to reduced SN-38 glucuronidation, which results in a long SN-38 half-life and increased area under the curve (AUC), causing late-onset diarrhea. UGT1A1 expression is highly variable. Clinical evidence indicates that the steady-state dosage of SN-38 can range up to 50-fold between UGT1A1*28 homozygous and heterozygous groups. Patients have a 5- to 9-fold greater risk of grade 4 leukopenia with a standard dosage of irinotecan. The FDA recommends dose reduction in patients with the homozygous UGT1A1*28 allele. Caution and possible dose reduction for patients with the heterozygous UT1A1 *28 allele is also advised (TABLE 2).5,6,21,22,35
CYP450: The PGx activity of CYP450 biomarkers (CYPs) plays a critical role in regulating the activation and inactivation of many chemotherapeutic agents (TABLE 1). Several of these agents are prodrugs and need to be activated by CYPs. CYP2B6 is one of the key enzymes involved in the activation of cyclophosphamide, ifosfamide, procarbazine, and thiotepa. Polymorphism of CYP2B6 may potentially affect the plasma level of active metabolites for these drugs and impact the drug efficacy and toxicities. Although over 50 polymorphisms of CYP2B6 have been identified, the most common functional variants are CYP2B6*5 and CYP2B6*6, with decreased gene expression and enzyme activity of CYP2B6. Takada et al reported higher rates of nephrotoxicity associated with CYP2B6*5 polymorphism in lupus patients treated with pulse cyclophosphamide.36 However, the role of CYP2B6*5 is still not clearly defined.4,11-13,31,32,36
G6PD and Rasburicase: Rasburicase, a recombinant form of urate oxidase, is approved by the FDA for the initial management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, or solid tumor cancers who are receiving anticancer therapy expected to result in tumor lysis syndrome. Rasburicase is usually well tolerated; however, several adverse reactions are of particular concern. The drug is contraindicated in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency (e.g., patients of African or Mediterranean ancestry). The enzyme activity is easily tested from a blood sample of the patient. Genetic testing can be performed to check mutations within families. G6PD deficiency is the most common disease-producing enzymopathy in humans. It is a metabolic enzyme involved in the pentose phosphate pathway, which is important in red blood cell metabolism, and is inherited as an X-linked recessive disorder, which affects 400 million people worldwide. The disease is highly polymorphic, with more than 300 reported variants. The excess peroxide poses a risk for both hemolytic anemia and methemoglobinemia. Hemolysis occurred in <1% patients who received rasburicase, with severe hemolytic reactions presenting within 2 to 4 days of the start of rasburicase.37,38
Biomarkers Related to Drug Transporters
Genetic polymorphisms of drug transporters are key contributors to multidrug resistance (MDR) to cancer treatments, which may lead to decreased efficacy and unpredictable toxicities associated with the therapy. The key players of MDR are a group of transporters known as adenosine triphosphate (ATP)-binding cassette (ABC) and human organic cation transporter (hOCT1), both of which play a critical role in drug efflux, especially for chemotherapeutic agents. There are seven families of ABC transporters named alphabetically from A to G, among which three ABC efflux pumps are particularly important for chemoresistance, including ABCB1, ABCC1, and ABCG2. P-glycoprotein (P-gp) belongs to ABCB1 and is the most widely studied drug transporter for MDR in chemotherapy due to its being a major obstacle for efficiency and safety. ABC transporters are ubiquitously expressed in many normal tissues and are overexpressed in tumor cells. For example, epithelial cells of the GI and biliary tract and normal hematopoietic stem cells express ABCB1 and ABCG2. ABCB1 and ABCC1 expression has been shown in gastrointestinal stromal tumor (GIST), while ABCB1, ABCG2, and OCT1 were found in mononuclear cells in chronic myelogenous leukemia (CML) patients. Interestingly, it has been proved that one of the revolutionary CML treatment agents (imatinib) is an inhibitor of ABCG2. Imatinib is also a substrate for hOCT1. However, it is still not clear whether hOCT1 polymorphism is associated with resistance to imatinib in CML.39
Overexpression of P-gp in tumor cells will allow the efflux of a large variety of structurally unrelated cancer drugs, such as corticosteroids and paclitaxel, decrease oral bioavailability of the drugs, and increase toxicities. Clinical evidence indicates that polymorphisms in ABC drug transporters may contribute to the varying individual responses to standard cancer treatment regiments. Over 48 SNPs of the ABCB1 gene have been identified. Hoffmeyer et al demonstrated that the polymorphism of the 3435T variant is associated with decreased expression of P-gp.40 Illmer et al reported the association of P-gp polymorphisms with complete remission, but not overall survival, in acute myeloid leukemia (AML).41 Furthermore, Kao et al reported that the polymorphism of P-gp overexpression contributes to the resistance to paclitaxel in non–small cell lung cancer (NSCL).42 In addition, Robey et al used cytotoxicity assays to prove that cancer cells transfected with mutant ABCG2 were more resistant to anthracyclines (doxorubicin, daunorubicin, epirubicin), compared to a wild-type ABCG2 control. ABCG2 is also known as an ABC transporter breast-cancer resistance protein (mitoxantrone resistance-associated protein).43
Biomarkers Related to Drug Targets
More and more evidence demonstrates that genetic polymorphism of drug targets not only plays an important role in drug efficacy and drug resistance, it may also lead to drug toxicity for cancer therapy. The epidermal growth factor receptor (EGFR) is constitutively expressed in many normal epithelial tissues. Overexpression of EGFR is also detected in many human cancers, including head and neck, colon, and rectal. EGFR is one of the most important therapeutic targets for colorectal cancer. EGFR gene overexpression is a strong indicator to predict the response to cetuximab, erlotinib, gefitinib, and panitumumab. Cetuximab and panitumumab are blocking monoclonal antibodies to EGFR. Binding of cetuximab and panitumumab to the EGFR on tumor cells has been shown to block receptor phosphorylation and activation of receptor-associated kinases, resulting in inhibition of cell growth, induction of apoptosis, and decreased matrix metalloproteinase and vascular endothelial growth factor production. In contrast, erlotinib and gefitinib directly inhibit the intracellular phosphorylation of the tyrosine kinase associated with the EGFR. Skin toxicities are commonly associated with therapeutic agents targeting EGFR.44,45
In a prospective clinical study, Graziano et al investigated the association of EGFR polymorphisms with overall survival, drug response, and toxicity for patients with a cetuximab-irinotecan regimen after failing a first-line FOLFOX regimen (5-FU, leucovorin, oxaliplatin).46 They reported that metastatic colorectal cancer patients with EGFR intron 1 short allele polymorphisms have higher risk of grade 2-3 skin toxicity compared with patients who carried the long alleles.46
Folate antagonists (methotrexate, pemetrexed, pralatrexate) and thymidylate synthase inhibitors (5-FU, capecitabine) are two important groups of chemotherapeutic agents.5,10 Methylenetetrahydrofolate reductase (MTHFR) is an enzyme that directs the folate pool toward homocysteine remethylation at the expense of DNA and RNA synthesis. Polymorphism in MTHFR (C677T) causes reduction in activity. This polymorphism has been reported in all populations worldwide (24%-40% in Caucasians, 26%-37% in Japanese, 11% in African Americans, 6.6% in Africans). This polymorphism leads to increased toxicity in patients receiving antifolate therapy, such as methotrexate and 5-FU. In one study, patients who had low levels of MTHFR activity experienced grade 4 toxicities with a chemotherapy regimen of cyclophosphamide, methotrexate, and 5-FU. More studies need to be conducted in order to properly define the relationship with MTHFR polymorphism and toxicities induced by antifolate therapy.4,33,34
Biomarkers Related to DNA Repairing and Detoxifying
Oxaliplatin is a third-generation platinum agent that is used in both adjuvant and metastatic settings. It is believed to form cross-links between complementary DNA strands, which result in DNA damage and apoptosis. Neurotoxicity is one of the primary toxicities of oxaliplatin. Excision repair cross-complementing group 1 (ERCC1) is an endonuclease that removes damaged DNA segments and is responsible for the rate-limiting process of nucleotide excision repair (NER). NER is a pathway that can result in platinum resistance–based cancer treatment, such as oxaliplatin, as it removes the cross-link that is formed by this drug.15,47 Preliminary data suggest that ERCC1-118 T/T is associated with increased ERCC1 expression. Higher levels of ERCC1 protein will result in less oxaliplatin toxicity due to an increased level of DNA repair. However, ERCC1-118 T/T is associated with shorter survival in patients with colorectal cancer and NSCLC treated by platinum drugs.48
GST enzymes are a family of enzymes responsible for detoxification of many chemotherapeutic agents and environmental toxins, including carcinogens. Polymorphism of GST, especially at the promoter region, has been associated with decreased GST enzyme activity. However, the decreased detoxifying activity associated with GST polymorphism may also cause increased toxicities associated with chemotherapy, such as cyclophosphamide- or platinum-based therapy. The GSTP1-105*G allele is associated with increased development of neurotoxicity in patients receiving FOLFOX-4 chemotherapy.14,48
Conclusion
One of the biggest challenges in oncology is to manage the toxicities associated with chemotherapy. Accrued evidence demonstrates that PGx biomarkers related to DMEs, drug transporters, drug targets, and drug detoxifying mechanisms play critical roles in predicting the safety, toxicity, and efficacy of cancer therapy in individuals or groups of patients. With clinically important biomarker polymorphisms becoming increasingly delineated, the need for clinicians to understand the association of these polymorphisms with potential changes of pharmacokinetics and pharmacodynamics of certain cancer therapies has become crucial. The integration of PGx biomarkers in chemotherapy offers clinicians the ability to individualize treatment in order to better predict and manage drug toxicities, improve quality of life, and maximize the efficacy of therapy.
REFERENCES
1. Albertini L, Siest G, Jeannesson E, Visvikis-Siest S. Availability of pharmacogenetic and pharmacogenomic information in anticancer drug monographs in France: personalized cancer therapy. Pharmacogenomics. 2011;12:681-691.
2. McLeord HL, Krynetski EY, Relling MV, et al. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia. 2000;14:567-572.
3. Ma Q, Lu A. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacological Rev. 2011;63:437-459.
4. Lee W, Lockhart AG, Kim RB, et al. Cancer pharmacogenomics: powerful tools in cancer chemotherapy and drug development. Oncologist. 2005;10:104-111.
5. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530-2535.
6. Hoskins JM, Goldberg RM, Qu P, et al. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99:1290-1295.
7. Gamelin E, Delva R, Jacob J, et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2099-2105.
8. Schwab M, Zanger UM, Marx C, et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131-2138.
9. Evans WE, Hon YY, Bomgaars L, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293-301.
10. Gor PP, Su HI, Gray RJ, et al. Cyclophosphamide-metabolizing enzyme polymorphisms and survival outcomes after adjuvant chemotherapy for node-positive breast cancer: a retrospective cohort study. Cancer Research. 2010;12:2-10.
11. Ekhart C, Doodeman VD, Rodenhuis S, et al. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics. 2008;18:515-23.
12. Rodriguez-Antona1 C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25:1679-1691.
13. Aebi S, Davidson T, Gruber G, Cardoso F; ESMO Guidelines Working Group. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011;22(suppl 6):vi12-vi24.
14. Ansari M, Lauzon-Joset JF, Vachon MF, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 2010;45:261-267.
15. Hoffmann AC, Wild P, Leicht C, et al. MDR1 and ERCC1 expression predict outcome of patients with locally advanced bladder cancer receiving adjuvant chemotherapy. Neoplasia. 2010;12:628-636.
16. Gréen H, Söderkvist P, Rosenberg P, et al. Pharmacogenetic studies of paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol. 2008;104:130-137.
17. Correia C, Vicente AM. Pharmacogenetics of risperidone response and induced side effects. Personalized Med. 2007;4:271-293.
18. Tomalik-Scharte D, Lazar A, Fuhr U, Kirchheiner J. The clinical role of genetic polymorphisms in drug-metabolizing enzymes. Pharmacogenomics J. 2008;8:4-15.
19. Ulrich CM, Robien K, McLeod HL. Cancer pharmacogenetics: polymorphisms, pathways and beyond. Nat Rev Cancer. 2003;3:912-920.
20. Camptosar (irinotecan) injection. Safety labeling changes approved by FDA CDER–May 2010. FDA Medwatch. www.fda.gov/Safety/MedWatch/
21. Bhushan S, McLeod H, Walko CM. Role of pharmacogenetics as predictive biomarkers of response and/or toxicity in the treatment of colorectal cancer. Clin Colorectal Cancer. 2009;8:15-21.
22. Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333:1171-1175.
23. Nilotinib. PharmGKB. www.pharmgkb.org/drug/
24. Omura K. Clinical Implications of dihydropyrimidine dehydrogenase (DPD) activity in 5-FU based chemotherapy: mutations in DPD gene, and DPD inhibitory fluoropyrimidines. Int J Clin Oncol. 2003;8:132-138.
25. Collie-Duguid ES, Etienne MC, Milano G, et al. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics. 2000;10:217-223.
26. Innocenti F, Iyer L, Ratain MJ. Pharmacogenetics: a tool for individualizing antineoplastic therapy. Clin Pharmacokinet. 2000;39:315-325.
27. Relling MV, Hancock ML, Boyett JM, et al. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817-2823.
28. Sachse C, Brockmoller J, Hildebrand M, et al. Correctness of prediction of the CYP2D6 phenotype confirmed by genotyping 47 intermediate and poor metabolizers of debrisoquine. Pharmacogenetics. 1998;8:181-185.
29. Tan SH, Lee SC, Goh BC, Wong J. Pharmacogenetics in breast cancer therapy. Clin Cancer Res. 2008;14:8027-8041.
30. CYP2D6 pharmacogenomics of tamoxifen treatment. Technol Eval Cent Asses Program Exec Summ. 2008;23:1-32.
31. Rohrbacher M, Kirchhof A, Geisslinger G, Lötsch J. Pyrosequencing-based screening for genetic polymorphisms in cytochrome P450 2B6 of potential clinical relevance. Pharmacogenomics. 2006;7:995-1002.
32. Wang H, Tompkins LM. CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Curr Drug Metab. 2008;9:598-610.
33. Schwahn B, Rozen R. Polymorphisms in the methylenetetrahydrofolate reductase gene: clinical consequences. Am J Pharmacogenomics. 2001;1:189-201.
34. Ulrich CM, Yasui Y, Storb R, et al. Pharmacogenetics of methotrexate: toxicity among marrow transplantation patients varies with the methylenetetrahydrofolate reductase C677T polymorphism. Blood. 2001;98:231-234.
35. Toffoli G, Cecchin E, Corona G, et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061-3068.
36. Takada K, Arefayene M, Desta Z, et al. Cytochrome P450 pharmacogenetics as a predictor of toxicity and clinical response to pulse cyclophosphamide in lupus nephritis. Arthritis Rheum. 2004;50:2202-2210.
37. Browning LA, Kruse JA. Hemolysis and methemoglobinemia secondary to rasburicase administration. Ann Pharmacother. 2005;39:1932-1935.
38. Beutler E. G6PD deficiency. Blood. 1994;84:3613-3636.
39. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105-127.
40. Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473-3478.
41. Illmer T, Schuler US, Thiede C, et al. MDR1 gene polymorphisms affect therapy outcome in acute myeloid leukemia patients. Cancer Res. 2002;62:4955-4962.
42. Kao CH, Hsieh JF, Tsai SC, et al. Quickly predicting chemotherapy response to paclitaxel-based therapy in non-small cell lung cancer by early technetium-99m methoxyisobutylisonitrile chest single-photon-emission computed tomography. Clin Cancer Res. 2000;6:820-824.
43. Robey RW, Honjo Y, Morisaki K, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971-1978.
44. Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040-2048.
45. Hecht JR, Patnaik A, Berlin J, et al. Panitumumab monotherapy in patients with previously treated metastatic colorectal cancer. Cancer. 2007;110:980-988.
46. Graziano F, Ruzzo A, Loupakis F, et al. Pharmacogenetic profiling for cetuximab plus irinotecan therapy in patients with refractory advanced colorectal cancer. J Clin Oncol. 2008;26:1427-1434.
47. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307-320.
48. Ruzzo A, Graziano F, Loupakis F, et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J Clin Oncol. 2007;25:1247-1254.
To comment on this article, contact rdavidson@uspharmacist.com.


