ABSTRACT: Pituitary tumors (adenomas) occur in approximately 17% of the U.S. population.1 Pituitary adenomas constitute 10% to 25% of all intracranial tumors, the majority of which are benign.2,3 Two theories have been formulated regarding the cause of pituitary adenoma development: intrinsic abnormalities versus hormonal stimulation.4,5 Recent technological advances in molecular biology, however, indicate that pituitary tumors arise from a single cell mutation followed by clonal expansion.5 Causes and risk factors are unique to each of the tumor types identified in this article; therefore, selection of the appropriate methods of diagnosis and treatment remains paramount.
Pituitary tumors, or adenomas, are common neoplasms, occurring in approximately 17% of the U.S. population.1 Currently, pituitary adenomas account for 10% to 25% of all intracranial tumors.2 The majority of these tumors are benign, and the development of cancer is rare.3
Overview
Because of its ability to secrete hormones that control many glands throughout the body, the pituitary gland is sometimes known as the “master endocrine gland.”2 This gland is a pealike structure that is located below the hypothalamus and above the sphenoid bone.6 Surrounded by the sella turcica—a bony, saddle-shaped structure—the pituitary gland consists of an anterior lobe and a posterior lobe. The posterior lobe is responsible for the release of oxytocin and antidiuretic hormone (ADH), whereas the anterior lobe synthesizes and releases six hormones: prolactin (PRL), adrenocorticotropic hormone (ACTH), growth hormone (GH), thyroid-stimulating hormone (TSH), luteinizing hormone (LH), and follicle-stimulating hormone (FSH).6 Abnormal cell growth in the pituitary gland constitutes a pituitary adenoma.2 Smaller adenomas, or microadenomas, are 1 cm or less; larger adenomas, or macroadenomas, are greater than 1 cm.2 Adenomas may be graded radioanatomically as follows: <1 cm, no sella expansion (grade 1); ≥1 cm, possible sella expansion (grade 2); ≥1 cm, invasion of floor or suprasellar extension (grade 3); and ≥1 cm, destruction of sella (grade 4).2
Signs, Symptoms, and Complications
Pituitary adenomas generate physical and biochemical changes. The size of the adenoma can influence the symptoms experienced. General and physical signs and symptoms include nausea, vomiting, vertigo, headache, impaired vision, runny nose (cerebrospinal fluid [CSF] leak), confusion, and seizures. Specific signs and symptoms are associated with certain types of pituitary tumor (TABLE 1).3,5,7-9
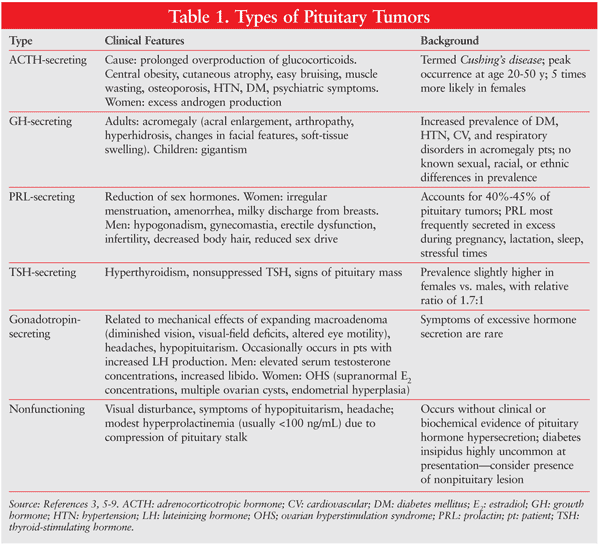
Because of the pituitary gland’s proximity to the regions of the brain, adenoma-associated morbidities include stroke, visual loss, meningitis, CSF leak, and cranial palsy.10 If the adenoma is diagnosed early, prognosis can be excellent, with surgery for total tumor removal carrying a mortality rate of less than 1%.10
Pathophysiology
There have been two theories concerning the origin of pituitary tumors. One hypothesis was that pituitary adenomas represented intrinsic abnormalities within the gland itself; the other theory was that adenomas originated from continuous stimulation by a hypothalamic hormone or factor.5 However, recent technological advances in molecular biology have shown that pituitary tumors arise from a single cell mutation followed by clonal expansion. Pituitary neoplasia is a multistep process involving dysregulation of cell growth, cell differentiation, and hormone production.5
Causes and Risk Factors
Whereas some pituitary tumors are idiopathic, others are linked to a rare genetic disorder, multiple endocrine neoplasia type 1 (MEN1). MEN1 can cause hyperactivity or enlargement of various endocrine glands, including the pituitary gland. Carney complex, another genetic cause of pituitary adenomas, is characterized by spotty skin pigmentation and tumors of connective tissue and the endocrine glands. While each of the aforementioned may cause pituitary tumors, other factors, such as increasing age and certain genetic conditions, may place patients at increased risk for developing pituitary tumors and therefore must also be considered.11
Diagnosis
Hormone measurement and imaging studies are the best methods for diagnosing pituitary adenomas.9 In addition, diagnostic tests that assess the hypersecretion of hormones can aid in the establishment of a differential diagnosis or the confirmation of a pituitary adenoma.
An unusually high prolactin level is almost exclusively associated with prolactinomas, but moderate elevation may occur during pregnancy or with the use of medications such as metoclopramide.7 In GH-secreting adenomas, an elevation of insulin-like growth factor 1 (IGF-1) is the most reliable indicator.7 The oral glucose tolerance test assesses GH levels after administration of glucose.12 Insufficient GH suppression after glucose administration is indicative of pituitary dysfunction.7 The release of IGF-binding protein-3 (IGFBP-3), which binds to IGF-1, depends upon the presence of GH.12 A determination of IGFBP-3 can be useful in diagnosing patients with normal levels of IGF-1 and GH.12
The diagnosis of ACTH-secreting adenomas relies primarily on cortisol elevation, which often is detected during preliminary testing.7 Excessive cortisol release may be investigated by conducting 24-hour urinary free cortisol, low-dose dexamethasone-suppression, or late-night salivary cortisol tests.7 The differential diagnosis of Cushing’s disease involves evaluation of plasma ACTH levels, high-dose dexamethasone-suppression tests, and corticotropin-releasing hormone stimulation tests.7 The most reliable tool for differential diagnosis is measurement of ACTH via venous drainage of the pituitary gland.7 TSH-secreting adenomas are characterized by increased levels of TSH, which will eventually trigger symptoms of hyperthyroidism.7 Hyperthyroidism due to TSH-secreting tumors may be distinguished from other causes of the condition by little to no TSH response during thyrotropin-releasing hormone stimulation and a high alpha subunit/TSH ratio.7 Hypersecretion of hormones is rare in cases of FSH- and LH-secreting tumors; however, free beta-FSH or alpha subunit secretion has been observed in some cases.7 For all types of pituitary adenoma, MRI is the recommended imaging method for assessing the tumor’s size and location.7,13
Nonpharmacologic Treatment
Treatment of a pituitary tumor depends upon several factors, including tumor type and size and the rate and extent of progression into the brain.3 Tumor removal via transsphenoidal surgery or craniotomy is required if the tumor is affecting other physiological structures, such as the optic nerve. In transsphenoidal surgery, an incision is made under the upper lip or at the base of the nose between the nostrils to allow the surgical instruments to reach the pituitary gland via the sphenoid bone.2 An endoscope is used for viewing purposes during the procedure, and a curette is used to remove the tumor tissue on the pituitary gland. One advantage of this type of surgery is the re-establishment of normal function in the pituitary gland with no visible facial scarring. Transsphenoidal surgery is not appropriate for tumors that have invaded nearby nerves or brain tissue.3
In a craniotomy, a more invasive incision is made in the upper part of the skull to facilitate the removal of a portion of the skull to reveal parts of the brain.3 The invasive nature of this procedure is a factor in some of the adverse effects associated with the surgical removal of pituitary adenomas.10
Radiation may be used to prevent tumor cells from replicating.3 The method of radiation depends upon the tumor’s characteristics. In external radiation, a machine beams radiation at the abnormal growth, whereas internal radiation involves placement of a radioactive substance directly into or near the growth. In stereotactic radiation, a head frame attached to the patient’s skull delivers a large dose of radiation directly to the tumor.2
The type of tumor diagnosed determines which surgical procedure is performed (TABLE 2).

Pharmacologic Treatment
Gonadotroph Adenomas: Standard therapy for gonadotroph adenomas has not been established. Most published data are based on case reports and anecdotal experience.8 Suppression of the secretory activity of the adenoma occasionally is accompanied by improvements in visual-field defects or tumor shrinkage. Attempts at treating nonfunctioning pituitary tumors with dopamine agonists (i.e., bromocriptine and cabergoline) and somatostatin analogues have been unsuccessful, partly because only a minority of these tumors have dopamine or somatostatin receptors. Bromocriptine typically has little effect on tumor size, but shrinkage has been reported in a few cases. Similarly, octreotide has been successful at improving vision in some patients, but the response is varied, and this drug should be reserved for therapeutically unresponsive patients. Consequently, there is no effective medical treatment for gonadotropin-secreting tumors.8
TSH-Secreting Pituitary Adenomas: TSH-secreting pituitary adenomas are rare, accounting for 1% to 2% of all pituitary tumors.5,14 Many TSH-secreting adenomas are stage III macroadenomas.7 The goal of treatment is to establish a state of euthyroidism. Patients with these tumors who display hyperthyroid symptoms warrant medical treatment. Great success has been observed with somatostatin analogues, namely octreotide.7 Octreotide is also formulated as a long-acting injection, which has been shown to increase cure rates by 80% to 90% compared with other therapies.5 Octreotide normalizes TSH levels in 79% of diagnosed patients and reduces tumor size in 52%.7 In high doses, dopamine agonists have been used to treat TSH-secreting pituitary tumors, but they are less effective than octreotide, perhaps because of abnormalities in adenoma dopamine receptor function.5
GH-Secreting Pituitary Adenomas: The goal of treatment for GH-secreting adenomas is to normalize GH and IGF-1 while preserving pituitary function. The initial treatment is transsphenoidal surgery.7 Ninety percent of microadenomas and 50% of macroadenomas are biochemically cured after resection.7 For patients with mediocre outcomes, therapy with a somatostatin analogue is the next option, based on proven efficacy. Sandostatin, which is typically reserved for postsurgical adjuvant therapy, is frequently used preoperatively in patients with cardiovascular or respiratory complications; it also is used to prevent perioperative complications due to elevated GH levels.7
Pegvisomant, a GH analogue, may be utilized for tumors that secrete GH. It blocks the effects of GH, but does not inhibit GH production or tumor size.8 Dopamine agonists offer only limited results for GH-secreting pituitary tumors; however, these agents are particularly useful in the treatment of pituitary tumors that cosecrete PRL and GH, with higher dosages required for efficacy.5
Prolactinomas: Bromocriptine and cabergoline are the drugs of choice for treating prolactinomas.2 These agents have been shown to effectively decrease prolactin levels and reduce tumor size in about 75% of patients, possibly leading to restoration of gonadal function.7 The initial dosing of bromocriptine is 1.25 to 2.5 mg per day. The dosage must be increased slowly to prevent nausea and dizziness.15 Gradual increases of 2.5 mg per day over a period of 2 to 7 days are recommended. Each dose should be taken with a meal or bedtime snack.15 The initial dosing of cabergoline (0.25 mg) is twice per week.15 The dosage must be titrated gradually, with increases made every 4 weeks.15 In addition to nausea and dizziness, common adverse effects noted with these agents include vomiting, constipation, postural hypotension, and nasal congestion.2
Cabergoline has a number of therapeutic advantages over bromocriptine.7 For example, cabergoline has a significantly longer half-life, which allows it to be administered once or twice per week, while bromocriptine must be administered daily.7 Cabergoline is reported to have less frequent and less severe side effects and is effective against tumors that are resistant to bromocriptine.7,15 However, bromocriptine is less expensive and has longer market tenure than cabergoline.15 Additionally, bromocriptine has a more extensive history of investigation in pregnant patients, making it the drug of choice during pregnancy.7
Although dopamine agonists are effective for correcting hypogonadism, other options, such as hormone replacement therapy, serve as an alternative for restoring reproductive function.7 Furthermore, recent studies have suggested that temozolomide, based on its favorable safety profile and significant reduction of hormone secretion and tumor size, could be a new option for treating aggressive prolactinomas.16
ACTH-Secreting Tumors: The treatment of ACTH-secreting tumors includes the utilization of drugs that inhibit adrenal cortisol secretion. The drug of choice, ketoconazole, is usually initiated at 200 mg by mouth two or three times daily, with a recommended maintenance dosage of 400 mg by mouth two or three times daily.5,17 Dosage adjustments are determined by monitoring 24-hour urinary free cortisol levels.17 The major adverse effect associated with high doses of ketoconazole is hepatic impairment, so liver function must be regularly monitored during treatment.7 Other therapeutic options to suppress cortisol secretion include metyrapone, aminoglutethimide, mitotane, and etomidate (TABLE 3).7

Conclusion
The pituitary gland secretes hormones that control various glands throughout the body. Abnormal cell growth within this structure can result in hormonal imbalances, which are a major cause of morbidity associated with pituitary adenomas. Given the multiple types of pituitary tumors that can arise, it is crucial in the diagnostic stage to observe the patient’s signs and symptoms and to measure hormone levels. Current surgical and pharmacologic treatment, coupled with close monitoring, may reduce the incidence of complications of pituitary adenomas, leading to increased progression-free survival.
REFERENCES
1. Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613-619.
2. National Cancer Institute. Pituitary tumors treatment (PDQ).
http://cancer.gov/cancertopics/pdq/treatment/pituitary/HealthProfessional.
Accessed December 15, 2011.
3. Mayo Clinic. Pituitary tumors. www.mayoclinic.com/health/pituitary-tumors/DS00533. Accessed December 15, 2011.
4. American Cancer Society. Pituitary tumors.
www.cancer.org/Cancer/PituitaryTumors/DetailedGuide/pituitary-tumors-what-causes.
Accessed March 25, 2012.
5. Arafah BM, Nasrallah MP. Pituitary tumors: pathophysiology, clinical manifestations and management. Endocr Relat Cancer. 2001;8:287-305.
6. Disorders of endocrine function. In: Porth CM, ed. Essentials of Pathophysiology: Concepts of Altered Health States. 2nd ed. Philadelphia, PA: Lippincott,Williams & Wilkins; 2006:673-697.
7. Freda PU, Wardlaw SL. Clinical review 110: diagnosis and treatment of pituitary tumors. J Clin Endocrinol Metab. 1999;84:3859-3866.
8. Buhk JH, Jung S, Psychogios MN, et al. Tumor volume of growth
hormone-secreting pituitary adenomas during treatment with pegvisomant: a
prospective multicenter study. J Clin Endocrinol Metab. 2010;95:552-558.
9. Johns Hopkins Medicine. Pituitary adenomas.
www.hopkinsmedicine.org/neurology_neurosurgery/specialty_areas/pituitary_center/pituitary-tumor/types/pituitary-adenoma.html.
Accessed December 28, 2011.
10. Johns Hopkins Medicine. Radiotactic surgery.
www.radonc.jhmi.edu/radiosurgery/disorders/pituitary.html. Accessed
January 22, 2011.
11. Cancer.Net. Carney complex. www.cancer.net/patient/Cancer+Types/Carney+Complex. Accesssed December 30, 2011.
12. Klibanski A, Ho K, Freda PU, et al. State-of-the-art strategies for the diagnosis and management of acromegaly. Endocrinologist. 2001;11:223-232.
13. Katznelson L. The diagnosis and treatment of acromegaly. Endocrinologist. 2003;13:428-434.
14. Brucker-Davis F, Oldfield EH, Skarulis MC, et al.
Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid
hormone sensitivity, and treatment outcome in 25 patients followed at
the National Institutes of Health. J Clin Endocrinol Metab. 1999;84:476-486.
15. National Endocrine and Metabolic Diseases Information Service.
Prolactinoma.
www.endocrine.niddk.nih.gov/pubs/prolact/prolact.aspx#treatment.
Accessed December 27, 2011.
16. Raverot G, Sturm N, de Fraipont F, et al. Temozolomide treatment
in aggressive pituitary tumors and pituitary carcinomas: a French
multicenter experience. J Clin Endocrinol Metab. 2010;95:4592-4599.
17. Gross BA, Mindea SA, Pick AJ, et al. Medical management of
Cushing disease. Medscape Today. www.medscape.com/viewarticle/566310_2.
Accessed December 29, 2011.
To comment on this article, contact rdavidson@uspharmacist.com.


