US Pharm. 2014;39(6)Generic suppl):13-20.
The Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act, is a federal law passed in 1984 that encourages the manufacture and market entry of generic pharmaceuticals.1 Prior to 1984, antibiotics already had an abbreviated generic approval process in place under section 505 of the Federal Food, Drug, and Cosmetic (FD&C) Act.2 In 1997, the Food and Drug Administration Modernization Act made antibiotics eligible for patent term restoration under the Hatch-Waxman Act.3
The Hatch-Waxman Act
The original intent behind the Hatch-Waxman Act was to increase generic drug availability in the marketplace and subsequently stimulate price competition to make drugs more affordable.4 However, it was also recognized that increased competition from generic manufacturers would decrease the financial returns for innovator companies, thereby reducing the incentive to discover and market new drug products. The Act sought to achieve the objectives of increasing competition with the introduction of a greater number of generic drugs in a timely manner and of fostering discovery by rewarding innovators with increased patent terms during which they would enjoy market exclusivity.5
The Hatch-Waxman Act amended the FD&C Act and created the Abbreviated New Drug Application (ANDA) process to submit generic drugs to the FDA for approval.6 In order for the FDA to approve an ANDA, one of four conditions, referred to as paragraphs, must be met (TABLE 1).6 Prior to the passage of the Act, companies seeking approval for a generic version of a drug had to independently conduct trials to prove safety and efficacy of their product, similar to the trials conducted by innovators. These trials resulted in additional, significant expenses for generic manufacturers similar to those of the innovator. As these generic companies would likely see diminished profits when compared to the innovator product due to the presence of a competing product immediately upon market entry, the incentive to introduce a generic was markedly less than that for a brand-name product. Therefore, as part of the ANDA process outlined in the amendment, generic drug formulations are considered therapeutically equivalent (referred to as bioequivalent) if the release pattern, drug concentrations, strength, purity, and formulation are comparable to those of the brand-name product.6

Bioequivalence studies are far less expensive and time-consuming to conduct, thus increasing potential profit margins and incentive to enter the market with a generic product. Furthermore, the first manufacturer with an approved ANDA under a paragraph IV certification may gain a 180-day generic exclusivity period in which the FDA will not approve additional generic products, thus increasing the price for which the first approved generic can be sold because of decreased competition.1
In order to balance the increased competition from generic manufacturers with the need to promote innovation and new drug discovery, the Act also incorporates an extension to the patents of innovator products. Most patents granted in the United States have a 20-year term beginning on the patent filing date (of note, at the time the Hatch-Waxman Act was passed, drug patents had a 17-year term after patent approval). However, since the patent approval process, a New Drug Application (NDA) submission, FDA approval, and initial manufacture of the product all occur after the original patent-filing date, drug companies marketing branded drugs typically do not realize a full 20-year patent-protected marketing term. To compensate brand-name drug manufacturers for the loss of marketing exclusivity due to the FDA approval process, the Act specifies that an innovator product may receive a patent term extension consisting of two parts. The first component of this extension is equal to half of the time between the initiation of human clinical trials and the submission of the NDA (known as the investigational new drug, or IND, period), and the second component is the entirety of the NDA review period. Both of these periods are added to determine the patent term extension. The Act also specifies that the patent extension is limited to either 5 additional years or a total effective patent protection of 14 years (whichever is less) .1
Breakthrough Therapy
Expedited drug pathways provide additional avenues for drug manufacturers to get products to market more quickly and are intended to facilitate the availability of innovative medications that address the unmet medical needs of serious or life-threatening conditions.7 ,8 Three long-standing FDA approaches exist: fast track, accelerated approval, and priority review.9,10 It has been almost 2 years since the newest pathway, breakthrough therapy designation, was introduced through the enactment of Section 902 and the Food and Drug Administration Safety and Innovation Act (FDASIA) on July 9, 2012.11 Unique to the breakthrough therapy designation, drug manufacturers now have the ability to work collaboratively with FDA managers and reviewers to expedite development for drugs that are intended alone or in combination with one or more drugs to treat a serious or life-threatening disease or condition.12 To be eligible for the breakthrough therapy designation, preliminary clinical evidence must exist indicating that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, surrogate markers, or pharmacodynamics biomarkers. The FDA states that these different designations are intended to be used in conjunction with one another to facilitate expedited development of new drugs.7 ,8
Although the significant benefits that breakthrough therapy potentially contributes to patients’ health should not be minimized, the accompanying benefits to drug companies should also be examined. A typical clinical drug-development program takes about 7 years and $1 billion to provide sufficient safety and efficacy data for FDA approval.13 However, under the breakthrough therapy designation, drugs may be approved nearly 3 years earlier than standard drugs as a result of the ability to file an NDA based on compelling phase II trial data, versus the phase III data typically required for a standard drug review.
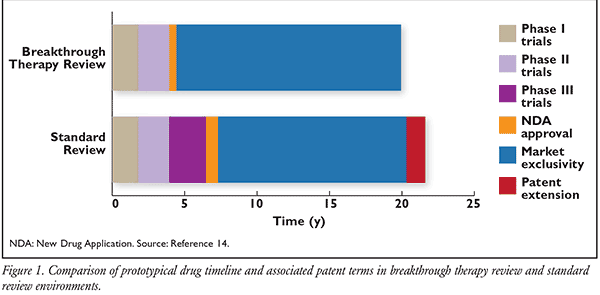
To help visualize this concept, FIGURE 1 compares a prototypical drug approved under both standard and breakthrough therapy designations.14 Standard drugs have an average of <13 years of effective patent protection remaining after approval, but could benefit from a 1.3-year patent extension under the Hatch-Waxman Act to a 14-year effective patent life. The same drug reviewed as breakthrough therapy would have about a 15.5-year effective patent life after approval, and would therefore not qualify for any patent term restoration under the Hatch-Waxman Act. As a result of decreased time in the FDA approval process, a drug reviewed as breakthrough therapy may generate an additional 3 years’ worth of sole-source revenue when compared to a standard drug. While these timelines are merely examples and may vary greatly based on many factors, the implications for the generic drug market from increased patent length of innovator products, increased manufacturing profitability, and approval of secondary indications are yet to been seen.
The Hatch-Waxman Act has demonstrated the extent to which market trends can be influenced by encouraging competition. The proportion of generic products used in the U.S. has steadily increased to over 80% in 2012,15 while research has continued to flourish. In less than 20 years, the first drugs approved under the breakthrough therapy designation will see their patents expire. The pressures they face and the cost to patients and the healthcare system to obtain them will be influenced not only by future research in respective treatment areas, but also by laws passed in the interim to regulate generic competition and patent terms of these unique medications.
The breakthrough therapy designation exists to put potentially life-saving treatments on the market and into the hands of patients more quickly. On one hand, scientists and drug manufacturers posit that due to improvements in the science and molecular biology of diseases, they are able to create more efficacious medicine with decreased toxicities.16 They argue that the FDA approval process for these safer medications that address major health problems should reflect the urgency and potential benefit these drugs provide. However, as a result, the manufacturers enjoy a quicker, cheaper path to an exclusive market consisting of patients with very few options—but this does not guarantee patients access to medications.
Despite a lack of phase III trials and the security that accompanies them, many of these medications are still exorbitantly expensive. For example, sofosbuvir ( Sovaldi)—an oral hepatitis C treatment recently approved under the breakthrough therapy designation—costs $84,000 for a complete 12-week course of therapy.17 Necessarily so, there is concern that this designation system is being abused, saturated, and not used as originally intended. This is easily recognized within the first year that Section 902 was introduced, with 94 total requests for breakthrough therapy designation being received by the FDA (32 of which were granted).13 TABLE 2 lists some of these drugs.18,19 It appears evident that expedited review processes such as the breakthrough therapy designation and fast track or priority review are providing a roundabout way for manufacturers to get their brand-name drugs to market in a more cost-effective and expeditious manner, and with a longer duration of marketing exclusivity.

Discussion
The Hatch-Waxman Act was also initially constructed to ensure that patients are receiving safe medications, as evidenced by extensive safety clinical trials. As the breakthrough therapy designation leads to drug approval based on smaller or shorter clinical trials, the possibility for unknown significant adverse events drastically increases. Consider that for standard drugs, significant safety warnings are found to emerge at a median of 11 years after approval.20 This is for drugs that have successfully passed the necessary phase II and III safety trials and does not recognize breakthrough drugs that are approved by clinical trials with smaller population sizes and/or shorter durations. Furthermore, because these medications may be used in such specific, small populations, it is possible that any safety concerns may not emerge until the drug has become generic and a larger population has been affected. Effectively, the immediate period after release of these agents is serving as a phase III clinical trial; meanwhile, the manufacturer reaps the benefit of market exclusivity. This delay in discovery of potential safety hazards may create concern for generic manufacturers if diligent pharmacovigilance has not occurred during brand-name product market exclusivity.
Given the unique timeline for approval of these drugs and the associated uncertainty when compared with standard approval processes, an argument can be made to decrease patent exclusivity on breakthrough therapies. By their very nature, these drugs treat hard-to-manage, complex, serious conditions and are therefore afforded a purchasing audience with little to no choice in treatment. This is in stark contrast to many drugs approved through standard means—those that must also provide more evidence and enter the market competing with other therapeutic options. If drug manufacturers themselves are publicly expressing that they are able to create more efficacious medicine with decreased toxicity and therefore should be able to bypass particular clinical trials (thus circumventing the expenditure for these trials) ,16 they should not be granted the same market exclusivity afforded to standard medications (or more, as the case may be).
The way to expand competition, make drugs more affordable, and increase options for patients with serious conditions with few treatment options is to recognize the advantages already associated with therapy and adjust patent terms to reflect them. By reducing marketing exclusivity and patent protection periods on drugs approved via the breakthrough therapy designation, we can pass along the benefits of a quicker time to market to patients in the form of expanded treatment options and additional generic competition leading to lower prices, rather than to the brand-product manufacturers in the form of additional profits.
Conclusion
Ultimately, the Hatch-Waxman Act aims to make drugs that are effective, safe, and affordable available to patients. As we venture into a new realm of targeted drug therapies deemed breakthroughs with the promise of treating previously difficult-to-manage conditions, pharmaceutical legislation is in need of a breakthrough—a set of laws and classifications as unique as the drugs they govern.
REFERENCES
1. Drug Price Competition and Patent Term Restoration Act of 1984. Public Law 98-417. www.gpo.gov/fdsys/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf. Accessed April 29, 2014.
2. §505 of the Federal Food, Drug, and Cosmetic Act of 1938. FD&C Act Chapter V: Drugs and Devices. www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/. Accessed April 29, 2014.
3. Food and Drug Administration Modernization Act of 1997. Public Law 105-115. www.gpo.gov/fdsys/pkg/PLAW-105publ115/pdf/PLAW-105publ115.pdf. Accessed April 29, 2014.
4. Mossinghoff G. Overview of the Hatch-Waxman Act and its impact on the drug development process. Food Drug Law J. 1999 ;54:187-194.
5. McGough KJ. Preserving the compromise: the plain meaning of Waxman-Hatch market exclusivity. Food Drug Cosmet Law J. 1990 ;45:487-504.
6. Wheaton JJ. Generic competition and pharmaceutical innovation—the Drug Price Competition and Patent Term Restoration Act of 1984. Cathol Univ Law Rev. 1986 ;35:433-487.
7. Fast track, breakthrough therapy, accelerated approval and priority review. FDA: For Consumers. December 28, 2013. www.fda.gov/forconsumers/byaudience/forpatientadvocates/speedingaccesstoimportantnewtherapies/ucm128291.htm. Accessed January 5, 2014.
8. Guidance for Industry. Expedited programs for serious conditions—drugs and biologics [draft guidance]. FDA. June 2013. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM358301.pdf. Accessed January 5, 2014.
9. Sherman R, Shapley S, Robb M, Woodcock J. Expediting drug development—the FDA’s new “breakthrough therapy” designation. N Engl J Med. 2013 ;369:1877-1880.
10. Frequently asked questions: breakthrough therapies. FDA. December 31, 2013. www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticAct-FDCAct/SignificantAmendmentstotheFDCAct/FDASIA/ucm341027.htm. Accessed January 5, 2014.
11. §902 (Breakthrough Therapies) of the Food and Drug Administration Safety and Innovation Act. July 9, 2012. www.gpo.gov/fdsys/pkg/BILLS-112s3187enr/pdf/BILLS-112s3187enr.pdf. Accessed April 29, 2014.
12. Dolgin E. First breakthrough drugs designated, but dilution worries linger. Nat Med. 2013 ;19:116-117.
13. Sellers A. How to maximize on FDA’s breakthrough therapy designation in your brand communications while you still can. PM360. October 28, 2013. www.pm360online.com/how-to-maximize-on-fdas-breakthrough-therapy-designation-in-your-brand-communications-while-you-still-can/. Accessed January 5, 2014.
14. DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003 ;22:151-185.
15. IMS Institute for Healthcare Informatics. Declining medicine use and costs: for better or worse? A review of the use of medicines in the United States in 2012. May 2013. http://static.correofarmaceutico.com/docs/2013/05/20/usareport.pdf. Accessed January 5, 2014.
16. Colliver V. Breakthrough drugs receive expedited FDA review. San Francisco Chronicle. August 6, 2013. www.focr.org/8-6-2013-san-fransisco-chronicle-breakthrough-drugs-receive-expedited-fda-review. Accessed January 6, 2013.
17. U.S. Food and Drug Administration approves Gilead’s Sovaldi ( sofosbuvir) for the treatment of chronic hepatitis C. December 6, 2013. www.gilead.com/news/press-releases/2013/12/us-food-and-drug-administration-approves-gileads-sovaldi-sofosbuvir-for-the-treatment-of-chronic-hepatitis-c. Accessed April 29, 2014.
18. NDA and BLA approval reports. Breakthrough therapy approvals. FDA. March 10, 2014. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/NDAandBLAApprovalReports/ucm373418.htm. Accessed April 29, 2014.
19. Breakthrough therapy—orphan drugs. www.orphan-drugs.org/wp-content/uploads/sites/4/2014/01/Capture7.png. Accessed April 29, 2014.
20. Moore TJ, Furberg CD. Development times, clinical testing, postmarket follow-up, and safety risks for the new drugs approved by the US Food and Drug Administration. JAMA Intern Med. 2013 ;174:90-95.
To comment on this article, contact rdavidson@uspharmacist.com.



