US Pharm. 2015;40(6)(Generic Drug suppl):22-29.
ABSTRACT: The use of biological products has increased exponentially over the last several decades. New regulations allow for the submission of a Biologics License Application (BLA) for a biosimilar or interchangeable biological agent to a reference product. Although the FDA has only approved one biosimilar product to date, this is expected to be a growing market in the United States. The Purple Book is an important compendium of FDA-approved biological products and their biosimilar and interchangeable products. This reference guide will assist pharmacists in providing the leadership necessary to develop and implement appropriate pharmacovigilance programs for biological products.
The number and complexity of biological products used in the management of human diseases have continued to rise since the introduction of growth hormone and insulin. Biological products in current therapeutic use include monoclonal antibodies, blood derivatives, and vaccines, as well as recombinant products such as cytokines, thrombolytics, and enzymes.1 Biological products are licensed with a product exclusivity period that is often 12 years from the first date of the agent’s first licensure.2
The exclusivity period of many of the currently available biological products on the market will end in the very near future. The expiration of the biological agent exclusivity period will be accompanied by the introduction and use of many biosimilar biological products to be the first reference biological agent in the market. Unlike small molecule drugs, biological products have large complex proteins with differences between protein structure and amino acid sequence.3 These differences in protein structure can induce antibody formation and trigger an immune response in the patient, resulting in every-thing from lack of desired clinical effect or outcome to induction of rare, serious, and life-threatening complications.
This article will discuss the FDA’s compendium of biological products, known as the Purple Book: Lists of Licensed Biological Products With Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations, and how pharmacists can use this guide to perform their role in the pharmacovigilance of biological and biosimilar products.
Biological Products: Manufacturing, FDA Approval, and Licensure
Several major steps are involved in the manufacturing of biological and biosimilar products. These production steps include modifying the selected gene of interest; inserting the gene in a specified cell line or host for the purpose of replicating the cell line and increasing protein expression; harvesting the protein products from the cell line; and purifying the selected protein.4 There are complexities and difficulties associated with all aspects of the production stages in the manufacturing process. These factors may impact the quality of the produced products, including complex and large molecules with complex three-dimensional structures, use of cell systems for production, and variation in the biological products due to changes during protein production.4 These production difficulties often translate into profound variation in both intra- and interindividual responses and complications in the use of biological agents.
Biological and biosimilar products are FDA-approved and licensed under the Public Health Services Act (PHSA), while small molecule drugs are FDA-approved and licensed under the Federal Food, Drug, and Cosmetic (FD&C) Act.5,6 A Biologics License Application (BLA) submitted under section 351(a) of the PHSA is approved and licensed after appropriate efficacy and safety data have been evaluated.5 A BLA submitted under section 351(k) of the PHSA is approved for licensure after appropriate demonstration of its similarity and absence of clinical meaningful difference to a 351(a) product, considered the referenced biological product, has been demonstrated. The exclusivity period for the reference product essentially ensures that a 351(k) application may not be submitted until 4 years after the first licensure of the reference product, and the FDA may not approve a 351(k) application until 12 years after the licensure of the reference product.7
The FDA’s Purple Book
The Purple Book is a compendium of FDA-approved biological products and their biosimilar and interchangeable products.8 It is similar to the Orange Book, which is a listing of approved generic drugs with therapeutic equivalency to brand products. Information for each product listed in the Purple Book includes its BLA tracking number, product name, product proprietary name, date of licensure, date of first licensure, reference product exclusivity expiration date, indication as to whether the product is interchangeable (I) or biosimilar (B), and whether the product was withdrawn from the market.8
- A reference biological agent denotes a product defined under section 351(i)(4) of the PHSA as the single biological product licensed by the FDA under section 351(a) of the PHSA against which the biosimilar biological agent was evaluated in its application submitted under section 351(k) of the PHSA.7
- A biosimilar agent is a product described under section 351(i)(2) of the PHSA as highly similar to the reference agent. A biosimilar agent that is licensed by the FDA under section 351(a) of the PHSA has been demonstrated to have no clinical differences between the biosimilar and the reference biological agent with regards to potency, purity, or safety.7
- An interchangeable biological agent indicates a product defined under section 351(k)(4) of the PHSA as a product licensed by the FDA under section 351(k) of the PHSA that is biosimilar and predictably produces the same clinical outcome as the reference biological agent in a given patient. Additionally, a classification of interchangeability indicates that administering the product more than once to the same patient showed a similar efficacy and safety profile that remained unchanged or diminished even when administration was alternated between the biological agent and the reference product.9
Biosimilar Prescribing, Dispensing, and Interchangeability
The BPCI Act: The Biologics Price Competition and Innovation (BPCI) Act of 2009 was enacted as a part of the Patient Protection and Affordable Care Act of 2010.10 The BPCI Act amended the PHSA to create an abbreviated pathway for approval of biological products demonstrated to be biosimilar to or interchangeable with an FDA-licensed reference biological product. The BPCI Act is similar to the Abbreviated New Drug Application (ANDA) process established through the 1984 Hatch-Waxman amendment to the FD&C Act that created generic-drug program approvals for small molecule drugs.11 The intent of the law was to lower the cost of biologics and give patients access to crucial biological therapies that they needed and otherwise might not be able to afford due to high cost.
The BPCI Act allows for the submission of a BLA for a biosimilar or interchangeable biological agent to a reference product. It requires the bio-similar application’s sponsoring organization to submit evidence demonstrating that the biological product is biosimilar to a reference product; utilizes the same mechanism of action for the proposed indications of use; and uses the same route of administration, dosage form, and strength as the reference product. Furthermore, submitted evidence in the BLA must demonstrate that there are no clinically meaningful differences in safety, purity, and potency between the biosimilar product and the FDA-licensed reference product.10
Finally, the BPCI Act also allows the FDA to approve a biosimilar product with a designation that it is interchangeable with the FDA-licensed reference biologic.10 FDA-designated biosimilar products are biological products that are FDA-licensed and are highly similar to an already FDA-approved reference biological product. Furthermore, the biosimilar product must demonstrate no meaningful clinical differences with the reference product in terms of safety, purity, and potency, although inactive ingredients with minor clinical differences are acceptable. A biosimilar can only be approved for use for the same indications as the referenced product. However, approved indications for a biosimilar may be less, but not more than, all of the indications for the reference product. The requirements for prescribing biosimilars are like those of other medications in that the use of a proprietary or nonproprietary name is essential on the prescription.
The FDA definition of interchangeable biological product is one that is both biosimilar to the FDA-approved referenced product and is expected to produce the same clinical result as the reference product in any given patient.7 Furthermore, a biological product that is designated as interchangeable must show that if more than one administration to a patient is necessary, the safety and reduced efficacy risks of alternating or switching between use of the biological product and the reference product are no greater than the risks established for use of the reference product without alternating or switching. In addition, biosimilar products approved as interchangeable may be substituted for the reference product by the pharmacist without the intervention of the prescribing healthcare provider.
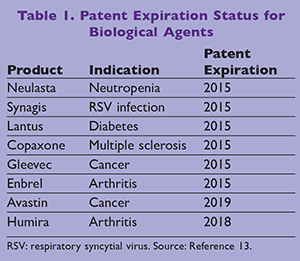
Under the BPCI Act, the FDA has approved only one biosimilar product and no interchangeable products thus far. Zarxio (filgrastim-sndz), a biosimilar product to Neupogen (filgrastim), was approved by the FDA in March 2015.12 Zarxio has the same indications as Neupogen, including febrile neutropenia in nonmyeloid and acute myeloid malignancy, severe neutropenic disorders, and radiation injury of bone marrow. However, it is not approved as an inter-changeable product and has not launched yet. Other biological products with patent losses between 2015 and 2019 for which there is a high likelihood of gaining approvals for biosimilars are shown on TABLE 1.13
Pharmacist’s Role in Biosimilar Pharmacovigilance
Biosimilar pharmacovigilance programs are essential to identifying, characterizing, and minimizing the risks associated with the use of biosimilars.4 There are multiple contributing risk factors associated with the use of biosimilars by patients. These risks are related to the nature of the manufacturing of the products, naming of the approved biologics, and associated errors that may result when the nonstandardized name is incorporated into the medical record and computerized drug codes. Biological agents have complex, three-dimensional structures produced from cell-based systems and may be more prone to generate products with potential variations due to minor changes that occur during manufacturing. These changes may trigger immunogenicity to the product when administered to patients. The consequences of immunogenicity of biologics include rendering the biological agent therapeutically and clinically ineffective through antibodies produced by the patient in neutralizing the administered biological agent. Patients may also have rare and serious, life-threatening autoimmune responses to the administered biosimilar product.
Pharmacovigilance programs for both biological and biosimilar products are necessary for monitoring postmarketing surveillance for the products.4 At a minimum, the pharmacovigilance program goal is to safeguard patient safety by detecting rare events associated with products and provide a mechanism to document and report problems. All healthcare providers have a duty to minimize risk to patients from the use of biological and biosimilar products. Thus, healthcare provider participation in a pharmacovigilance program is a professional responsibility. Effective communication among all healthcare providers is necessary to ensure the success of any pharmacovigilance program.
Pharmacovigilance programs include the FDA Adverse Event Reporting System (FAERS) through the MedWatch program, Risk Evaluation and Mitigation Strategies (REMS), and Risk Management Programs to reduce medication errors. The MedWatch program allows both healthcare providers and consumers to report serious adverse drug reactions and problems with drugs and medical devices to the FDA.14 In addition, manufacturers are also required to submit reports of adverse events to the FDA.
The goal of REMS is to provide clinicians with risk minimization strategies to ensure that the benefit of the prescription products outweighs any risks associated with their use.15 Each REMS has a specified safety measure unique to the safety risk of the product, which is also documented on the product label. No two REMS are the same. REMS require certain elements to ensure safe use, including that prescribers obtain special training or certification; the pharmacy or pharmacists be certified before dispensing the drug; dispensing is limited to specialized centers; and each patient is subject to monitoring and enrollment in registries. REMS for biological products include a medication guide, communication plan, elements of safe use, and an implementation system. Biological products with approved REMS are listed on the FDA website, and include such agents as Epogen/Procrit (epoetin alfa), Prolia (denosumab), Stelara (ustekinumab), and Tysabri (natalizumab).15
Overall, the goal of these programs is to reduce and eliminate medication errors and negative outcomes of drug use. The Institute of Medicine (IOM) and the Institute for Safe Medication Practices (ISMP) have tools to assist healthcare practitioners to develop and implement effective programs that help reduce medication errors.16,17
Pharmacists must play a leadership role in developing and implementing effective monitoring and reporting of adverse events to the FDA MedWatch and REMS programs in order to prevent and document medication errors. Pharmacists must also ensure correct attribution of all safety events to specific products. Programs must accurately document and effectively track what was offered versus what the patient received through electronic medical records and bar code administration. Finally, to ensure that pharmacovigilance is carried through the continuum of care, pharmacists must implement medication reconciliation programs that include transitions of care.
Conclusion
Approvals of complex biological products for the management of human diseases will continue to rise. It is expected that the numbers of biosimilar and interchangeable products will also increase. Pharmacists need to understand the complex nature of biological products, the approval process, the biosimilar and interchangeable designation of biological products, and their own part in the dispensing and use of these products in patients. The FDA’s Purple Book is a useful compendium that can assist pharmacists in their role in implementing pharmacovigilance programs for biological products.
REFERENCES
1. Simoens S. Biosimilar medicines and cost effectiveness. Clinicoecon Outcomes Res. 2011;3:29-36.
2. Barlas S. Senate may compromise on biosimilars: but length of market exclusivity poses a stumbling block. P T. 2008:33(8):442.
3. Renner L, Nkansah FA, Dodoo AN. The role of generic medicines and biosimilars in oncology in low-income countries. Ann Oncol. 2013;24(5):29-32.
4. Dranitsaris G, Amir E, Dorward K. Biosimilars of biological drug therapies: regulatory, clinical and commercial considerations. Drugs. 2011;71(12):1527-1536.
5. FDA. Federal Food, Drug, and Cosmetic Act (FD&C Act). www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm. Accessed March 8, 2015.
6. FDA. Public Health Service Act. www.fda.gov/RegulatoryInformation/Legislation/ucm148717.htm. Accessed March 8, 2015.
7. FDA. Biosimilars guidances. Updated April 28, 2015. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm290967.htm. Accessed May 1, 2015.
8. FDA. CDER list of licensed biological products. Updated March 6, 2015. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. Accessed May 1, 2015.
9. FDA. Guidance for industry on biosimilars: Q & As regarding implementation of the BPCI Act of 2009: questions and answers part I. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm259809.htm. Accessed March 8, 2015.
10. U.S. Congress. Section 7001-7003 (Biologics Price Competition and Innovation Act of 2009) of the Patient Protection and Affordable Care Act (Public Law 111-148). www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/ucm216146.pdf. Accessed March 8, 2015.
11. Drug Price Competition and Patent Term Restoration Act. Public Law 98-417. www.gpo.gov/fdsys/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf. Accessed March 8, 2015.
12. FDA. FDA approves first biosimilar product Zarxio. March 6, 2015. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed March 8, 2015.
13. The top 10 patent losses of 2015. FiercePharma. December 17, 2014. www.fiercepharma.com/special-reports/top-10-patent-expirations-2015. Accessed May 10, 2015.
14. FDA. MedWatch Online Voluntary Reporting Form. www.accessdata.fda.gov/scripts/medwatch/. Accessed March 8, 2015.
15. FDA. Approved Risk Evaluation and Mitigation Strategies (REMS). www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm111350.htm. Accessed March 8, 2015.
16. Institute of Medicine. Preventing medication errors: quality chasm series. July 20, 2006. www.iom.edu/en/Reports/2006/Preventing-Medication-Errors-Quality-Chasm-Series.aspx. Accessed March 9, 2015.
17. Institute for Safe Medication Practices. Medication safety tools and resources. www.ismp.org/tools/default.asp. Accessed March 9, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.



