US Pharm.
2008;33(6)(Generic Drug Review):30-34.
Pharmacists should be familiar
with the legal issues involved when they dispense a drug manufactured by a
company other than the one that makes the brand name of the prescribed drug.
This activity goes by many names: "drug product selection," "generic
substitution," "generic drug interchange," and probably a few others. For
simplicity, the term "generic substitution" will be used hereafter.
Pharmacists should also recognize the federal government's role in regulating
these activities. Finally, pharmacists must be familiar with the state laws
affecting these practices. While there is significant variation between the
different state laws, the laws can be categorized into just a few types. This
article will address each of these subjects.
Before proceeding, it is
essential to gain an understanding of some terminology. The terms important to
generic substitution are summarized in TABLE 1.1 The FDA
classifies as therapeutically equivalent products that are approved as safe
and effective; are pharmaceutical equivalents (i.e., contain identical amounts
of the same active drug ingredient in the same dosage form and route of
administration and meet compendial or other applicable standards of strength,
quality, purity, and identity); are bioequivalent (i.e., do not present a
known or potential bioequivalence problem and meet an acceptable in vitro, or
in some cases in vivo, or both, standard--or, if they do present such a
known or potential problem, are shown to meet an appropriate bioequivalence
standard); are adequately labeled; and are manufactured in compliance with
current Good Manufacturing Practice (GMP) regulations.2 Products
that meet these criteria are considered therapeutically equivalent even though
they may differ in certain other characteristics such as shape, scoring
configuration, release mechanisms, packaging, excipients (including colorings,
flavorings, and preservatives), expiration date/time, minor aspects of
labeling (e.g., presence of specific pharmacokinetic information), and storage
conditions. The FDA takes the position that when differences of these types
are important in the care of a particular patient, it may be appropriate for
the prescribing physician to require that a particular brand be dispensed
("dispense as written") as a medical necessity ("brand medically necessary").
With this limitation, however, the FDA believes that products classified as
therapeutically equivalent can be substituted with the full expectation that
the substituted product will produce the same clinical effect and safety
profile as the prescribed product.

Bioavailability testing is
used to determine whether one drug product has the same effect as a generic
equivalent. Bioequivalence can be determined through one of four mechanisms:
pharmacokinetic studies, pharmacodynamic studies, comparative clinical trials,
or in vitro studies. The last three of the aforementioned are used to test
drug products in which plasma levels are not affected, such as nasal sprays,
aerosols, and topical medications.3 Pharmacokinetic studies test
bioequivalence by measuring plasma levels to determine the rate and extent of
absorption. These tests are conducted with 24 to 36 volunteers, and single
doses of test and reference (or standard or innovator) drugs are administered.
If two drug products containing the same active chemical entity can reach the
site of absorption in similar times and be absorbed to the same extent,
bioequivalence can be established. The bioequivalence ranges measured by
pharmacokinetic parameters are between 80% and 125%.4 This means
that if the generic version of the reference product shows 20% less absorption
or up to 25% more absorption, the products will be deemed bioequivalent and,
in FDA terms, presumptively therapeutically equivalent.
Federal Regulations
Most pharmacists
already know that the Orange Book, created in 1980 and now in its 28th
edition, is an FDA publication that lists many drug products and contains
indications as to whether generic versions of medications are considered to be
"equivalent" to the drugs manufactured by the innovator company and most often
marketed with brand names. Pharmacists, however, might not recognize the
limitations inherent in the Orange Book evaluations. What is commonly
called the Orange Book is really entitled Approved Drug Products
with Therapeutic Equivalence Evaluations. (In the days when books were
published only in hardcover format, this text was bound with an orange cover,
hence its popular name; in these days of Internet publications, the online
version is usually referred to as the Electronic Orange Book.5
) One of the more significant limitations of this federal publication comes
directly from the first part of its formal name: Approved Drug Products.
The only drugs listed in this book are those that have been approved by the
FDA as so-called new drugs.6 The FDA does not list drugs
that are on the market without a new drug approval (NDA). Some drugs are still
being marketed in the United States without the benefit of an NDA because they
were on the market before the original Federal Food, Drug, and Cosmetic Act
(FDCA) took effect in 1938.7 These pre-1938 drugs were said to have
been "grandfathered," meaning that they could remain on the market without the
manufacturer's having to produce data showing that the drug was "safe" for its
intended purposes.8 In fact, the preface to the Orange Book
states: "Drugs on the market approved only on the basis of safety (covered by
the ongoing Drug Efficacy Study Implementation [DESI] review [e.g., Donnatal
® Tablets and Librax® Capsules] or pre-1938 drugs
[e.g., Phenobarbital Tablets]) are not included in this publication."
9 The DESI reference applies to drugs that were put on the market after
1938 but before 1962, when the FDCA was amended to require manufacturers to
submit to the FDA satisfactory data showing that their drug is effective (as
well as safe) for its intended purposes.10 Often called the Drug
Efficacy Amendment, these provisions also established GMP requirements and
significantly modified the clinical phases of drug testing.
It is important to note that
exclusion from the Orange Book does not imply that a drug is not safe
and effective or that it is not therapeutically equivalent to other similar
drug products. In point of fact, exclusion means only that the FDA has not
evaluated the safety, effectiveness, or quality of the excluded drug.11
The proper status of the
Orange Book contents as a legal source for pharmacists engaged in
generic-substitution decisions is somewhat complicated. The FDA makes special
note that the publication is not an official, legally binding regulation.
12 The FDA explicitly states that the listing of drugs with therapeutic
equivalency constitutes advice and that it does not mandate which drug
products may be prescribed, dispensed, or substituted for one another.13
It also draws a clear distinction between therapeutic-equivalence evaluations
and generic substitution. Equivalence evaluations "are a scientific judgment
based upon evidence, while generic substitution may involve social and
economic policy administered by the states, intended to reduce the cost of
drugs to consumers."14
State Laws
These disclaimers
aside, many states have elevated the Orange Book lists to legal status
by indicating that drugs the FDA deems to have equivalencies may be
substituted or, conversely, that drugs the FDA does not list as having
equivalencies cannot be substituted. All states in the U.S. have laws
addressing generic substitution to one degree or another. There are "positive
formulary" states, which identify generics that can be substituted, and there
are "negative formulary" states, which list drugs that cannot be substituted.
There are also states that do not refer to Orange Bookstandards and
have nether a positive nor a negative formulary, and where pharmacists are
permitted to perform generic substitution so long as the drugs are
pharmaceutically equivalent. See TABLE 2, which identifies which laws
apply in which states.15-17
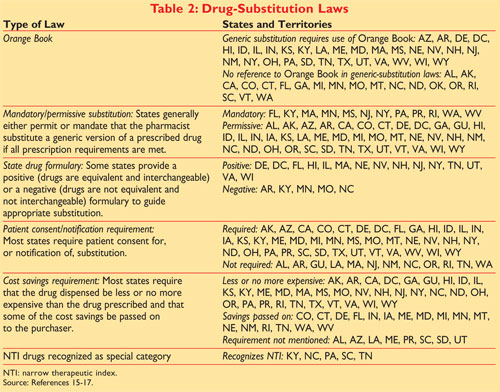
Some states have unique laws
about substituting drugs. In Oklahoma, for example, the law states that it is
unlawful for a pharmacist to substitute any like drug, medicine, chemical, or
pharmaceutical preparation without the authority of the prescriber or
purchaser.18 In Hawaii, it is unlawful for pharmacists to
substitute an equivalent generic product for any antiepileptic drug without
practitioner and patient consent.19 In Florida, each community
pharmacy must establish a formulary of generic and brand-name drug products
that, if selected as drug product of choice, would not pose a threat to the
health and safety of patients receiving prescription medication.20
In Iowa, the Board of Pharmacy advises use of the Orange Book to
determine therapeutic equivalency.21 The Kentucky Board of Pharmacy
maintains a list of narrow therapeutic index (NTI) drugs that cannot be
substituted, including digitalis glycosides, antiepileptic drugs,
antiarrhythmic agents, conjugated estrogens, esterified estrogens, warfarin
anticoagulants, theophylline products, and thyroid preparations.22
Likewise, the North Carolina Board of Pharmacy maintains a list of NTI drugs
that are not to be substituted. The 2007 list of NTI drugs includes:
carbamazepine (all oral dosage forms); cyclosporine (all oral dosage forms);
digoxin (all oral dosage forms); ethosuximide; levothyroxine sodium tablets;
lithium (all salts, all oral dosage forms); phenytoin (all salts, all oral
dosage forms); procainamide; theophylline (all salts, all oral dosage forms);
and warfarin sodium tablets.23 The Pennsylvania Board of Pharmacy
also maintains a list of NTI drugs that are not to be substituted.24
In South Carolina, the Board recommends not substituting three types
of medications, "1) including narrow therapeutic index drugs, e.g. lithium);
2) Premarin or Synthroid; and 3) ‘Critical drugs' in the following categories:
anticoagulants, anticonvulsants, anti-asthmatics (especially time-release
products), insulin, and cardiac glycosides."25 In Tennessee,
"a pharmacist may substitute A-rated products and use his judgment on unrated
products except for antiepileptic drugs used to treat patients with epilepsy
or seizures."26
It is interesting that certain
states recognize NTI drugs and have prohibitions, to some extent, about
substituting products in this category. Even more curious is that the FDA has
adopted the position since at least 1997 that NTI drugs do not need to be
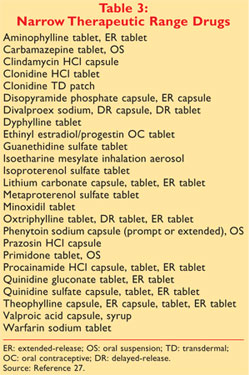
regulated any differently than other classes of drugs.27
Nevertheless, in 1995 the FDA developed a list of NTI drugs for purposes of
determining which types of studies should be done with specific dosage forms
to make bioequivalence determinations.28 The drugs identified by
the federal government as NTI drugs are given in TABLE 3. In 1990, the
Acting Commissioner of the FDA stated in a letter to the Pennsylvania
Department of Health that the FDA does not formally designate NTI drugs in the
Orange Book or elsewhere.
Conclusions
Pharmacists are
given significant responsibility by both federal and state government to help
control drug costs by making effective generic-substitution choices. Knowing
when not to perform generic substitution is an equally important task. The FDA
has taken much of the guesswork out of evaluating whether two drugs may be
substituted for one another with publication of the Orange Book. Not
all drugs legally marketed in this country are included in that text, however.
Nonetheless, unrated drugs may be substituted if the relevant criteria are
met. States take different approaches to the regulation of generic
substitution. Pharmacists must be familiar with the laws governing the states
where the practice of pharmacy is conducted.
REFERENCES
1. FDA. Approved
Drug Products with Therapeutic Equivalence Evaluations. 28th ed. 2008.
www.fda.gov/cder/orange/obannual.pdf. Accessed April 17, 2008.
2. Kazmi S. Pharmacy
practice: controversies in drug substitution. Medscape Pharmacists. November
7, 2007. www.medscape.com/viewarticle/563959_print. Accessed April 15, 2008.
3. Id.
4. See note 1, supra.
5. FDA. Electronic
orange book. Approved drug products with therapeutic equivalence evaluations.
www.fda.gov/cder/ob/. Accessed April 11, 2008.
6. §505 [21 USC 355] of
the Federal Food, Drug, and Cosmetic Act of 1938 (as amended).
www.fda.gov/opacom/laws/fdcact/fdcact5a.htm#sec505. Accessed April 11, 2008.
7. 52 Stat. 1040
(1938), 21 USC §301 et seq.
8. FDA. Guidance for
FDA Staff and Industry Marketed Unapproved Drugs--Compliance Policy Guide, Sec.
440.100, Marketed New Drugs Without Approved NDAs or ANDAs. Appendix A.
Brief history of FDA marketing approval requirements and categories of drugs
that lack required FDA approval. Revised June 2006.
www.fda.gov/cder/Guidance/6911fnl.htm#_Toc128533964. Accessed April 11, 2008.
9. FDA. Preface.
Approved Drug Products with Therapeutic Equivalence Evaluations. 28th ed.
2008. www.fda.gov/cder/ob/docs/preface/ecpreface.htm. Accessed April 11, 2008.
10. 76 Stat. 780 (1962).
11. Introduction. §1.5,
General Policies and Legal Status: x. Electronic Orange Book. See note 5,
supra.
12. "Therapeutic
equivalence evaluations in this publication are not official FDA actions
affecting the legal status of products under the Act." See note 5, supra.
13. See note 7, supra.
14. Id.
15. National
Association of Boards of Pharmacy. Survey of Pharmacy Laws. XIX: Drug
product selection laws. 2006.
16. State regulations
on generic substitution (as modified July 3, 2007). Pharmacist's Letter.
2006;22:220901.
www.pharmacistsletter.com/(S(grtd3055rt3i2cb0c14kjh23))/pl/ArticleDD.aspx?
nidchk=1&cs=FACULTY&s=PL&pt=2&dd=220901#STATE. Accessed April 15, 2008.
17. Christiansen TP,
Kirking DM, Ascione FJ, et al. Drug product selection: legal issues. J Am
Pharm Assoc. 2001;41:868-873.
18. OS 1961.
19. Department of
Commerce &Consumer Affairs. Hawaii revised statutes.
www.hawaii.gov/dcca/areas/pvl/main/hrs. Accessed April 15, 2008.
20. Online Sunshine.
The 2007 Florida statutes.
www.leg.state.fl.us/Statutes/index.cfm?App_mode=Display_Statute&URL=Ch0465/ch0465.htm.
Accessed April 15, 2008.
21. 2007 Iowa Code
Chapter 155A. Iowa Pharmacy Practice Act. www.state.ia.us/ibpe/pdf/IC155A.pdf.
Accessed April 15, 2008.
22. 201 KAR 2:116. Drug
products with therapeutic problems. www.lrc.state.ky.us/kar/201/002/116.htm.
Accessed April 15, 2008.
23. North Carolina
Board of Pharmacy. Pharmacy rules, statutes, and board policies.
www.ncbop.org/LawsRules/StatutesMay2007.pdf. Accessed April 15, 2008.
24. Commonwealth of
Pennsylvania generic drug equivalency/substitution laws & regulations.
http://ecapps.health.state.pa.us/pdf/ddc/generic33.ps.pdf. Accessed April 15,
2008.
25. South Carolina Code
of Laws (unannotated). www.scstatehouse.net/code/t39c024.htm. Accessed April
15, 2008.
26. See note 16, supra;
Public Acts, 2006. Chapter No. 866. Senate Bill No. 3738.
www.state.tn.us/sos/acts/104/pub/pc0866.pdf. Accessed April 15, 2008.
27. Therapeutic
equivalence of generic drugs: response to National Association of Boards of
Pharmacy. April 16, 1997. www.fda.gov/cder/news/ntiletter.htm. Accessed April
15, 2008.
28. Scale-up and
post-approval changes for intermediate release products (SUPAC-IR) guidance
document. Appendix A: Narrow therapeutic range drugs.
www.fda.gov/cder/guidance/cmc5.pdf. Accessed April 15, 2008.
To comment on this article, contact
rdavidson@jobson.com.



