US Pharm. 2013;38(7):69-79.
ABSTRACT: Stevens-Johnson syndrome (SJS) is a rare, life-threatening mucocutaneous reaction that is often drug-induced. SJS and toxic epidermal necrolysis (TEN) are considered to be the same condition on two ends of a spectrum, differing only by the extent of epidermal detachment. Despite reports of more than 100 drugs being taken in patients who develop SJS/TEN, a few classes such as antibiotics and anticonvulsants are thought to carry the highest risk. Genetic susceptibility due to polymorphisms of the human leukocyte antigen (HLA) gene may influence the development of SJS/TEN. There is conflicting evidence on the benefit of treatment of SJS/TEN with immunomodulating therapies. Although SJS is rare, pharmacists need to be prepared to identify and manage the acute and long-term effects of this condition.
Cutaneous (skin) reactions account for a large portion of drug-induced adverse effects. These reactions can range from a mild penicillin-induced rash to life-threatening conditions involving both the skin and mucous membranes, progressing to further organ involvement. One example of a life-threatening mucocutaneous condition is Stevens-Johnson syndrome (SJS). With the broad spectrum of potential severity, it is important for clinicians to be able to recognize the differences between cutaneous reactions, identify potential causative agents, and initiate therapy if indicated. The early recognition of severe cutaneous reactions is especially important to reduce fatal outcomes.
SJS is a mucocutaneous immunologic reaction that is often drug-induced and can result in long-term sequelae and mortality. The incidence of SJS is estimated at 1 to 6 cases per million person-years.1 SJS itself was first described in 1922 with the publication of a case report involving two young boys. The authors described the presence of extensive cutaneous eruptions with severe mucosal and ocular involvement, and stated that the condition was “unlike anything previously observed.”2 Other than case reports, most current knowledge of SJS comes from studies conducted in other countries, especially in Europe.
In 1956, a dermatologist named Alan Lyell described a mucocutaneous condition similar to but more severe than SJS, toxic epidermal necrolysis (TEN) or Lyell’s syndrome.3 Both SJS and TEN are characterized by epidermal detachment and erosive mucosal lesions. After years of debate, SJS and TEN are now considered to be the same condition but differ in severity and cutaneous involvement.4 The percentage of epidermal detachment is the primary differentiating factor between SJS and TEN, with SJS presenting with <10% epidermal detachment and TEN presenting with >30%.5
Both SJS and TEN are debatably included in the same spectrum as erythema multiforme (EM). This mucocutaneous condition has similarities in clinical presentation to SJS/TEN but has some distinct differences. TABLE 1 outlines some of the differences in the categorization of EM, SJS, and TEN.5,6 Disputes regarding the categorization of these conditions into the same spectrum revolve around the marked differences between SJS/TEN and EM in the common etiology and rate of recurrence. The most common cause of EM is the herpes simplex virus (HSV) infection, which contrasts with the medication-induced reaction in SJS/TEN.4,6 Due to the viral etiology in EM, recurrence can occur in 30% of patients; for SJS/TEN, recurrence is rare unless a patient is reexposed to the causative medication.6
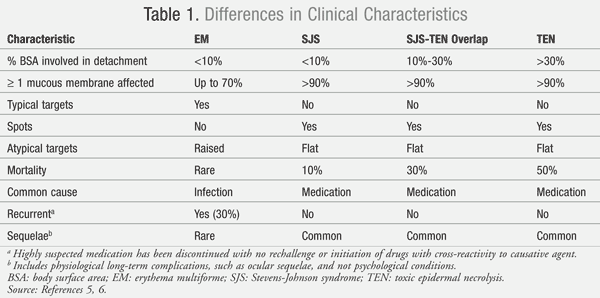
The appearance of the cutaneous lesions is another differentiating feature distinguishing EM from SJS/TEN. EM lesions have a target appearance with concentric rings of color and a dark center, while SJS/TEN lesions are irregularly shaped with a dark center that progressively coalesces.4-6 SJS/TEN patients normally present with flat macules (spots), which are absent in EM. Mortality is rarely seen in EM, while mortality in patients with SJS is about 10%.4-6
Clinical Presentation
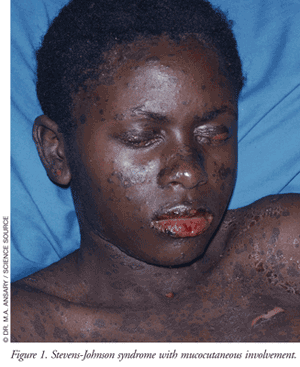
The typical clinical course of SJS begins within 8 weeks (usually 4 to 30 days) following the first exposure to the causative agent. Only in very rare cases where an inadvertent rechallenge occurs do symptoms appear within hours. Patients who develop SJS/TEN can have varying levels of cutaneous, extracutaneous, and mucous membrane manifestations (TABLE 2).4,7-9 About one-third of patients will present with nonspecific symptoms (e.g., fever, headache, sore throat, cough, malaise) and/or burning of the eyes followed by the appearance of mucocutaneous lesions of the eyes, mouth, genitals, and urinary tract in 1 to 3 days.4 Another third of patients will present with mucous membrane lesions (FIGURE 1), while the remainder of patients present with a diffuse rash. Following the appearance of the diffuse rash, the lesions convert to flaccid blisters, which spread with pressure and break easily, leading to extensive epidermal detachment. The detachment of the epidermis can be effortless, normally as a result of frictional trauma and on pressure points. A positive Nikolsky’s sign, which is the dislodgment of the epidermis with lateral pressure, may be present in these patients.4,7

Almost all patients with mucosal involvement develop painful hemorrhagic erosions coated by grayish white pseudo-membranes and crusts in the oral cavity and on the border of the lips.4 Involvement of the gastrointestinal (GI) tract can also affect the esophagus, small bowel, and colon, which may impact enteral nutrition and the absorption of oral medications.8 Ocular involvement is frequent, reported in up to 80% of patients, and can involve severe conjunctivitis and blepharitis along with visual disturbances and photophobia.4 Eyes can appear swollen, erythematous, and crusted as a result of ocular discharge.4, 9 Involvement of the respiratory tract epithelium may occur leading to hypoxemia, hypocapnia, and acid-base disturbances potentially requiring the need for mechanical ventilation.9 Patients who develop respiratory epithelium involvement have a higher risk of mortality. About 20% of TEN patients will develop epithelium involvement of the trachea and bronchi.9
Septicemia is another serious complication and is the most common cause of death in SJS/TEN. Normally, the epithelium provides a natural barrier to the systemic invasion of bacteria, but this barrier is compromised as large areas of the epidermis become detached. Extensive detachment can result in the collection of intravascular fluid in nontraditional body areas such as the peritoneal or pleural cavities, also known as third spacing of intravascular fluid and protein loss; this may lead to hypotension and possible organ dysfunction.8,9
The effects of SJS continue beyond the healing of the cutaneous lesions to include long-term complications such as vision loss, severe dry eye syndrome, GI/gynecological strictures, and nail disfigurement.4,7-9 Even though the cutaneous lesions rarely scar the skin, patients can exhibit hyper- and hypo-pigmentation of the affected areas.9 Low severity or lack of acute ocular symptoms is not predictive of the presence or absence of subsequent ocular sequelae.7 Survivors of this condition may display symptoms of posttraumatic stress disorder (PTSD) and have a fear of syndrome reoccurrence with exposure to a variety of medications.
Pathophysiology and Genetics
The specific pathophysiology of SJS and TEN is not well defined at this time. There are several theories as to the key immunologic players in their development, along with continued debate over whether it’s the parent drug or a metabolite that results in immune cell activation. The rare and seemingly random occurrence of SJS/TEN cannot be explained by medication exposure alone. Research into genetic susceptibilities has begun to elucidate the potential reasons why some individuals develop such a severe mucocutaneous reaction while others remain unaffected. Studies have shown that the presence of specific human leukocyte antigen (HLA) genotypes, which is a human version of the major histocompatibility complex (MHC), are associated with an increased risk of SJS/TEN when individuals are exposed to specific medications.10
Recent studies have identified a relationship between HLA-B*1502 and the development of SJS/TEN in people given carbamazepine who were of Southeast Asian descent (e.g., Han Chinese, Malaysian).11 The presence of HLA-B*1502 in the Han Chinese confers a 7.7% predictive value that a patient could develop carbamazepine-induced SJS/TEN, whereas the absence of this allele has a negative predictive value of 100%.11 This same increase in susceptibility was not demonstrated in European populations.11 Interestingly, the majority of patients in the European study who were positive for HLA-B*1502 and developed carbamazepine-induced SJS/TEN were of Southeast Asian descent.11,12 This ethnic selectivity may be explained by the fact that Europeans have a low prevalence of this particular HLA allele, 0.1%, compared to individuals with Southeast Asian ancestry (4.8%-12.8%), making the numbers needed to detect a statistically significant difference difficult to obtain.11 As a result, genetic testing for HLA-B*1502 is only recommended in patients of Southeast Asian descent.
HLA-B*5801 is another allele that has demonstrated a predictive relationship with a specific medication, allopurinol, and the development of SJS/TEN. In one study, all 51 patients who developed allopurinol-induced SJS/TEN were positive for HLA-B*5801 compared to only 15% (20/135) of allopurinol-tolerant patients.13 In Taiwan, the allopurinol labeling was recently updated to recommend genetic screening of patients prior to initiation of therapy.14 Currently, there is no FDA recommendation to test for HLA-B*5801 prior to therapy. Different from HLA-B*1502, this allele is equally distributed among ethnic groups, making genetic testing less practical.13
The development of the cutaneous lesions and epidermal necrosis are thought to occur as a result of massive apoptosis of keratinocytes. This is suspected to be a cell-mediated cytotoxic reaction. Studies have confirmed the presence of various cytotoxic cells, including natural killer T cells (NK) and drug-specific CD8+ T lymphocytes, within early cutaneous lesions.10 These cytotoxic cells are thought to lead to the amplification and release of cytokines, such as granulysin, perforin, and granzyme B, which likely play a separate role in apoptosis (FIGURE 2).4,10

Original theories proposed the interaction between Fas and Fas ligand (FasL), a member of the tumor necrosis factor (TNF) family found on the surface of activated T cells, as the main differentiating factor that leads to the extensive apoptosis of keratinocytes, cells of the epidermis, and the epidermal detachment not seen in other drug-mediated hypersensitivity reactions.8,10 The binding of FasL to Fas is known to induce activation of caspases, which are proteases that mediate apoptotic cell death, necrosis, and inflammation.4,10 This theory was challenged when recent studies identified elevated concentrations of granulysin in the blister fluid of patients with SJS/TEN.15 Unlike granulysin, the concentrations of granzyme B, perforin, and soluble Fas ligand (sFasL) in the blister fluid were insufficient to produce distinct cytotoxicity.15 Interestingly enough, the concentration levels of granulysin identified in the blisters directly correlated with the clinical severity of the symptoms; SJS lesions contained lower concentrations of granulysin compared to TEN lesions, which could explain the differences of clinical severity in these pathologically identical conditions.15 This relationship was further explored when granulysin was injected into mice and the clinical features of SJS/TEN, blistering and considerable epidermal and dermal necrosis, soon developed.15 Granulysin has a direct apoptotic effect on keratinocytes and is now thought to be a key mediator in the apoptosis of keratinocytes.7,10
Causes
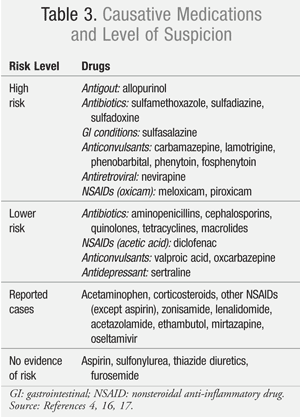
Up to 60% of cases of SJS can demonstrate causality to a medication exposure, but other factors including infection (e.g., Mycoplasma pneumonia) have been implicated in the development of this mucocutaneous condition, and up to 20% of cases remain idiopathic.4 More than 100 medications have been identified as potential causative agents of SJS/TEN. Even though some drugs have been implicated in case reports, not all of these agents have demonstrated a strong association with the development of SJS (TABLE 3).4,16,17 In two large European case-control studies, fewer than a dozen medications accounted for half of the analyzed SJS/TEN cases.16,17 Some of the most notorious medications associated with the development of SJS/TEN include sulfonamide antibiotics, antiepileptics (carbamazepine, phenytoin, lamotrigine, phenobarbital), allopurinol, nevirapine, and certain oxicam nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., meloxicam, piroxicam).16 One important distinction to note is that aspirin, unlike other NSAIDs, has not been associated with the development of SJS/TEN.
The presence of certain conditions, such as HIV, collagen vascular disease, and cancer can increase the risk of developing SJS.4 This is thought to be due to abnormalities of the immune system as a result of these conditions.
Current evidence is limited as to how significant the risk of cross-reaction is between structurally similar medications. Avoidance of these agents may not be a viable option in certain medical conditions. Seizure disorders can be particularly challenging due to structural similarities of several of the commonly used antiepileptics. Treatment with levetiracetam may be a viable option because of a lack of structural similarity with the high-risk anticonvulsants and with no strong association to the development of SJS/TEN. In a study by Locharernkul et al, some patients who developed either carbamazepine- or phenytoin-induced SJS were noted to have been exposed, and demonstrated tolerance, to other antiepileptics (phenytoin, phenobarbital, valproic acid, carbamazepine, and/or lamotrigine).18 Nine out of 10 patients with phenytoin- or carbamazepine-induced SJS/TEN in this study were at one point treated with valproic acid without incidence. These observational findings suggest that the risk of cross-reactivity may not be as significant as once thought, but more research is needed to confirm these findings.
Treatment and Management
The pharmacist can be an asset in the acute- and long-term management of SJS. Outpatient pharmacists are encouraged to emphasize the importance of reporting new rash symptoms in patients who are recently started on medications more commonly associated with SJS.
In the acute setting, prompt identification and removal of the highly suspected agent is vital. It is important to gather a detailed medication history in order to identify the most likely causative agent. The knee-jerk reaction to discontinue all of the patient’s medications is not appropriate and can complicate the clinical course if chronic conditions are not adequately treated. Each medication’s duration of therapy is important in implicating or excluding drugs most likely to be involved; the history should focus on those medications that were initiated within the last 8 weeks.4 If a causative agent is identified, rechallenge with that medication is not recommended, and if unintentionally done may cause a rapid recurrence of symptoms. Patients should be evaluated for future anxiety and potential PTSD symptoms when they are initiated on new medications.
Prompt initiation of appropriate treatment can potentially reduce the morbidity and mortality associated with SJS. Supportive care, similar to that provided to burn victims, is a vital component in the acute management of patients with SJS. Patients will commonly have fluid and electrolyte abnormalities that require careful monitoring; a recommended equation for fluid replacement is 0.7 mL/kg per percentage of body surface area (BSA) affected.9 Fluid requirements for SJS patients are normally 66% to 75% of those required in burn patients with the same extent of BSA involved. To prevent hypoperfusion of the kidneys, urine output should be maintained at 50 to 80 mL/h.7 Nonstick dressings can be placed on denuded areas of the body, with or without a saturated anti-infective, such as 0.5% silver nitrate.19 Topical anti-infectives with a sulfa moiety (e.g., silver sulfadiazine) should be avoided, especially if the causative agent is a sulfa derivative.9
Mouth care with disinfecting mouthwashes (chlorhexidine) and mild ointments (white petroleum) is essential in managing the mucosal lesions of the oral cavity and lips.9,19 Every patient, whether acute ocular involvement is apparent or not, should have his or her eyes evaluated, preferably by an ophthalmologist.9 Treatment can include prophylactic ophthalmic antibiotics (e.g., bacitracin or a fluoroquinolone), preservative-free emollients, antiseptic eye drops, and/or vitamin A. Recent evidence suggests that the use of ophthalmic topical steroids (fluorometholone ointment 0.1% every 1-2 hours for about 1-2 weeks) and amniotic membranes may help to preserve visual acuity and protect against scarring.20 Routine use of oral or parenteral prophylactic antibiotics is not recommended, although patients should frequently be monitored for the signs and symptoms of an infection and sepsis. Wound debridement is not always necessary because, unlike in burn victims, the epidermis can reepithelialize if it is maintained in place.7 Removal of the activated immune cells though plasmapheresis and hemodialysis has demonstrated mixed results in studies and is currently not considered the standard of care.7,21,22
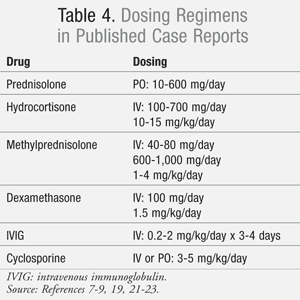
Intravenous immunoglobulin (IVIG) and corticosteroids are two therapies thought to improve clinical outcomes when used in addition to supportive care. The evidence to support the use of either IVIG or corticosteroids is conflicting and complicated by a wide variation in dosing and duration of therapy (TABLE 4).7-9,19,21-23 Whether to provide patients with high-dose pulse therapy or low-dose extended therapy is still being investigated.21 Cyclosporine is another immunomodulating therapy that has been investigated with varying results.9,23 There is currently no consensus as to what is the best medication regimen to reduce the clinical severity, complications, and occurrence of sequelae. If immunomodulating therapy is to be utilized, it should be initiated as soon as the diagnosis of SJS/TEN is determined. Currently, there are no prospective, randomized, placebo-controlled trials available to confirm whether either therapy reduces mortality or long-term sequelae. When data for 281 patients from the EuroSCAR study were retrospectively evaluated, no difference was found in mortality between IVIG and/or corticosteroids when compared to supportive care alone.24
Summary
SJS is a life-threatening mucocutaneous reaction that is commonly medication induced. Symptoms can be cutaneous, extracutaneous, or involve the mucous membrane and lead to further body system involvement if not addressed. Once the acute manifestations of the condition have healed, patients can experience long-term sequelae and psychological issues. Early identification and discontinuation of the causative agent are important. Currently there is no consensus on the use of various immunomodulating therapies for acute treatment of SJS. Future studies will hopefully address the ambiguous data that are currently available.
REFERENCES
1. Chan HL, Stern RS, Arndt KA, et al. The incidence of
erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal
necrolysis. A population based study with particular reference to
reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
2. Stevens AM, Johnson F. A new eruptive fever associated with stomatitis and opthalmia: report of two cases in children. Am J Dis Child. 1922;24:526.
3. Lyell A. Toxic epidermal necrolysis: an eruption resembling scalding of the skin. Br J Dermatol. 1956;68:355-361.
4. Valeyrie-Allanore L, Roujeau J. Chapter 40. Epidermal
necrolysis (Stevens-Johnson syndrome and toxic epidermal necrolysis).
In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
5. Bastuji-Garin S, Rzany B, Stern RS. Clinical
classification of cases of toxic epidermal necrolysis, Stevens-Johnson
syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92-96.
6. Roujeau J. Chapter 39. Erythema multiforme. In: L Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
7. Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:1-11.
8. Gerull R, Nelle M, Schaible T. Toxic epidermal necrolysis and Stevens-Johnson syndrome: a review. Crit Care Med. 2011;39:1521-1532.
9. Mockenhaupt M. The current understanding of Stevens-Johnson syndrome and toxic epidermal necrolysis. Expert Rev Clin Immunol. 2011;7:803-815.
10. Chung WH, Hung SI. Recent advances in genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci. 2012;66:190-196.
11. Fernando SL, Broadfoot AJ. Prevention of severe
cutaneous adverse drug reactions: the emerging value of pharmacogenetic
screening. CMAJ. 2009;182:476-480.
12. Lonjou C, Borot N, Sekula P, et al. A European study
of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis
related to five high-risk drugs. Pharmacogenet Genomics. 2008;18:99-107.
13. Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele
as a genetic marker for severe cutaneous adverse reactions caused by
allopurinol. PNAS. 2005;102:4134-4139.
14. Hershfield MS, Callaghan JT, Tassaneeyakul W, et al.
Clinical Pharmacogenetics Implementation Consortium guidelines for human
leukocyte antigen-B genotype and allopurinol dosing. Clin Pharmacol Ther. 2013;93:153-158.
15. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key
mediator for disseminated keratinocyte death in Stevens-Johnson
syndrome and toxic epidermal necrolysis. Nat Med. 2008;14:1343-1350.
16. Roujea JC, Kell JP, Naldi L, et al. Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. N Engl J Med. 1995;333:1600-1607.
17. Mockenhaupt M, Viboud C, Dunant A, et al.
Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of
medication risk with emphasis on recently marketed drugs. The EuroSCAR
study. J Invest Dermatol. 2008;128:35-44.
18. Locharernkul C, Loplumlert J, Limotai C, et al.
Carbamazepine and phenytoin-induced Stevens-Johnson syndrome is
associated with HLA-B*1502 allele in Thai population. Epilepsia. 2008;49:2087-2091.
19. Fromowitz JS, Ramos-Caro FA, Flowers FP, et al.
Practical guidelines for the management of toxic epidermal necrolysis
and Stevens-Johnson syndrome. Int J Dermatol. 2007;46:1092-1094.
20. Shammas M, Edward C, Sarkar J, et al. Management of
acute Stevens-Johnson syndrome and toxic epidermal necrolysis utilizing
amniotic membrane and topical corticosteroids. Am J Ophthalmol. 2010;149:203-213.
21. Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
22. Knowles S, Shear NH. Clinical risk management of Stevens-Johnson syndrome/toxic epidermal necrolysis spectrum. Dermatol Ther. 2009;22:441-451.
23. Huang YC, Li YC, Chen TJ. The efficacy of intravenous
immunoglobulin for the treatment of toxic epidermal necrolysis: a
systematic review and meta-analysis. Br J Dermatol. 2012;167:424-434.
24. Schneck J, Fagot JP, Sekula P, et al. Effects of
treatments on the mortality of Stevens-Johnson syndrome and toxic
epidermal necrolysis: a retrospective EuroSCAR study. J Am Acad Dermatol. 2008;58:33-40.
To comment on this article, contact rdavidson@uspharmacist.com.


