US Pharm. 2015;40(6):HS26-HS30.
ABSTRACT: Primary malignant intraocular neoplasms are relatively rare, but they can be fatal if not treated promptly. The two most common primary malignant intraocular cancers are uveal melanoma (in adults) and retinoblastoma (in children). Factors indicative of a poor prognosis include tumor size, ciliary-body involvement, epithelioid cells, extraocular extension, lymphocytic and melanophagic infiltration, mitotic activity, vascular-mimicry patterns, and the presence of monosomy 3 and a Class-2 expression profile in tumor cells. Depending upon the type and stage of eye cancer, treatment options include surgery, radiotherapy, laser therapy, chemotherapy (ChT), and targeted therapy. Eye-sparing therapies for retinoblastoma, including brachytherapy and systemic and intra-arterial ChT, have reduced the need for enucleation.
The term eye neoplasm refers to a cancerous growth in any part of the eye (eyeball, orbit, or adnexal structures).1 Eye cancers may be grouped into three basic categories according to their location: tumors of the eyelid and conjunctiva; intraocular tumors; and orbital tumors. Eye cancers are classified as primary (starts within the eye) or metastatic (spreads to the eye from another organ). The most common primary malignant intraocular tumor in adults is uveal melanoma (UM). The two most common cancers that spread to the eye from another organ are breast cancer and lung cancer; less common cancers include prostate, kidney, thyroid, skin, colon, lymphoma, and leukemia.2 The most common malignant intraocular tumor in young children is retinoblastoma. Given the number of eye-related cancers and their complexity, this overview will focus on the two most common primary malignant intraocular neoplasms: retinoblastoma and uveal melanoma.
Retinoblastoma
Most cells in this tumor histologically resemble retinoblasts (undifferentiated embryonic retinal cells).2,3 At a certain developmental point, these cells stop dividing and grow into mature retinal cells. Typically, this process is controlled by normal RB1 genes, but in rare cases, something goes wrong. Instead of maturing into special cells that detect light, some cells mutate, divide, and grow out of control, forming the cancer. This resemblance prompted Verhoeff to coin the term retinoblastoma, which was adopted by the American Ophthalmological Society in 1926 as a general term for this eye cancer.4
Retinoblastoma typically occurs in young children and can run in families. About 90% of cases are diagnosed in patients younger than 5 years of age.2,3 In some patients, both eyes are affected. The tumor starts in the retina, which is behind the pupil, in one or both eyes. These children are born with a mutation in the RB1 gene (germline mutation); that is, the RB1 gene is congenital in all cells in the body. These individuals usually develop retinoblastoma in both eyes, may develop several tumors within the eye, and are at risk for developing cancers in other body tissues as well. If retinoblastomas are not detected and treated, they can continue to grow and fill much of the eyeball.2,3
Uveal Melanoma
UMs can develop in any part of the uveal tract, including the iris, ciliary body, and choroid. Iris melanomas are less common than choroidal melanomas (approximately 5% vs. >80% of cases).5 Iris melanomas also have a better prognosis than ciliochoroidal tumors (10-year survival approximately 95% vs. 77%). Because of the less aggressive nature of iris melanomas, these tumors are usually managed conservatively (i.e., by close monitoring).2 Fast-growing tumors may be excised.
UMs are often referred to by their location; i.e., choroidal melanoma, ciliary-body melanoma, or iris melanoma. True iris melanomas originate within the iris, as opposed to originating elsewhere and invading the iris. Tumors arise from the pigment cells that color the eye (melanocytes) inside the uvea. Iris melanomas, although derived from uveal melanocytes, have more in common with cutaneous melanomas in that they frequently harbor BRAF mutations associated with ultraviolet (UV) damage.6,7 Mutations in the BAP1 gene are strongly linked to metastatic spread and patient survival.8 About 5% of UMs involve the iris.
The incidence of intraocular tumors varies markedly depending on the age and ethnicity of the patient population. UM preferentially affects the choroid of light-eyed, fair-skinned individuals of European descent (TABLE 1).1,2,9,10 A study conducted in New York City reported that the annual age-adjusted incidence (per million population) of UM was 0.31 in black patients.2 UM’s propensity for lightly pigmented persons and the posterior part of the uveal tract suggests that UV light may play a role in pathogenesis, but studies are inconclusive.11,12 Although retinoblastoma is the most common eye cancer worldwide, UM is the most common ocular cancer in Europe and the United States.6

Intraocular Melanoma
Intraocular melanoma preferentially affects the choroid of light-eyed individuals and develops within the eyeball and orbit.2,9,10 Melanomas develop from the pigment-making melanocytes. When melanoma develops in the eyeball, it is usually in the uvea. People with UMs have a 50% survival rate. Most people succumb to hepatic metastasis, which is unresponsive to current therapy. About nine out of 10 intraocular melanomas develop in the choroid or ciliary body of the uvea.9,10 Choroid cells make the same kind of pigment as skin melanocytes, so it is not surprising that these cells sometimes form melanomas.
The most commonly reported symptom of intraocular melanoma is an insidious and painless loss of vision. Other symptoms are a white pupil; worsening vision over weeks to months; blurred vision or sudden loss of vision; floaters or flashes of light; loss of part of the visual field; a growing dark spot on the iris; change in pupil size or shape; bulging of the eye; and change in how the eye moves.1,5,9 Some people with eye cancer do not experience any typical symptoms; others may think that their symptoms are caused by a medical condition other than cancer.2,9 Since patients with intraocular melanoma frequently have no related symptoms, often an ophthalmologist or optometrist detects the melanoma during the patient’s regular eye examination.
Oncogenic mutations in GNAQ or GNA11 (genes encoding for widely expressed G protein alpha subunits), which are observed in >80% of primary UMs, activate signaling pathways, including the mitogen-activated protein kinase pathway.13 Most other intraocular melanomas originate in the iris.2 They are easily seen because they often start in a dark spot on the iris. These melanomas usually grow fairly slowly, rarely spread to other parts of the body, and generally have a good prognosis. About 50% of UMs harbor mutations in the GNAQ or GNA11 genes that encode G protein–coupled receptors in the RAF/MEK/ERK pathway, also found in benign lesions. Congenital ocular melanocytosis GNAQ mutations are thought to be an early or initiating event in the pathogenesis of UM.2,14-16
In the U.S., UM is about seven times more common than retinoblastoma; retinoblastoma is three to four times more common than melanoma in India, and is also more common in Africa, South America, and parts of Asia. Intraocular melanomas are generally made of cells that are almost round, but have some straight edges. Most tumors are composed of two different kinds of cells: spindle cells and epithelioid cells. The prognosis is better if tumors are mostly spindle cells, as opposed to mostly epithelioid cells. Epithelioid tumors are more likely to metastasize to distant parts of the body, such as the liver.2
Changes in DNA and Genes
Changes in DNA and genes may be used to identify individuals at higher risk for developing eye cancers. It has been discovered that some families have a BAP1 gene mutation that renders them more likely to develop eye melanomas.1,2,10 These genetic changes in tumors may also help predict the likelihood of tumor spread. For example, in UM, the loss of one copy of chromosome 3 has been linked to an increased risk of spread. Also, patterns of gene expression appear to effectively predict whether an eye melanoma is likely to spread. Based on gene patterns, about one-half of eye melanomas can be classified as Class 1 tumors (low risk of spread); the rest fall into Class 2, which confers a high risk of spread.2
Survival Rates
The American Cancer Society estimates that 2,580 new eye melanomas will be diagnosed in the U.S. in 2015.1,10 The estimated number of cases is 1,360 in men and 1,220 in women. About 270 deaths (roughly 140 men and 130 women) from eye and orbital cancer are estimated.10 The National Cancer Institute’s Surveillance, Epidemiology, and End Results data on about 1,500 patients diagnosed with eye melanoma between 1988 and 2001 show that, overall, about 3 out of 4 people with eye melanoma survive for at least 5 years.17 Survival rates tend to be better for earlier-stage than for later-stage cancers, but accurate survival rates for eye melanomas based on a specific stage are hard to determine because these cancers are fairly rare.10,17,18 When the cancer is confined to the eye, the 5-year relative survival rate is about 80%.17 For people with eye melanomas that have spread to distant parts of the body, the 5-year relative survival rate is about 15%. If there is spread outside the eye, a stage is assigned (TABLE 2).18 Approximately 50% of patients with UM will develop metastases, usually fatal, by 10 to 15 years after diagnosis. The mortality rate is about 50% with or without treatment.2,10
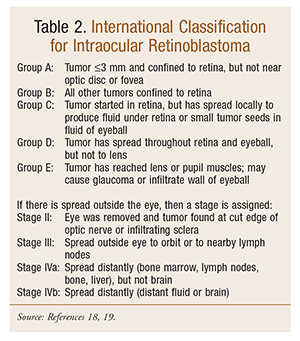
Eye Cancer Treatment
After an eye cancer is diagnosed and staged, treatment options include surgery, radiation therapy (RT), laser therapy, chemotherapy (ChT), and targeted therapy. The therapy selected depends on the type and stage of cancer, and in some instances, more than one of type of treatment may be used.2,3 General goals for treating intraocular melanoma are to reduce the risk of tumor spread and to maintain the health and vision of the patient’s eye. Cancer staging systems used for retinoblastoma include Reese-Ellsworth, International Classification for Intraocular Retinoblastoma (TABLE 2), and that of the American Joint Commission on Cancer.3,18-20 These staging systems can be used to establish the severity of retinoblastoma and guide the choice of an appropriate treatment plan. Selecting the best treatment options and/or recommendations depends on several factors, including tumor size, location, type and stage of cancer, possible adverse effects (AEs), and overall health. The following sections discuss several treatment approaches that may be considered.
Active Surveillance/Observation: This method is used if the intraocular melanoma is small or slow-growing and/or if treating the cancer would cause more discomfort than the disease itself, such as in patients without any symptoms and older or seriously ill patients. Active treatment may begin if the tumor shows signs of becoming more aggressive or spreading.2
Surgery: Parts of the eye or the entire eye may be removed, depending on the size and spread of the tumor. Surgical options include iridectomy (removal of part of the iris), iridocyclectomy (removal of the iris and ciliary body), sclerouvectomy/endoresection (removal of the choroidal tumor while preserving the eye), and enucleation (removal of the eye).2
Laser Therapy: Laser therapy uses focused beams of light to destroy eye tissue.2
RT: This therapy employs high-energy x-rays or other particles (i.e., external-beam radiation that uses protons rather than x-rays) to kill cancer cells.2
ChT: ChT uses various IV-administered drugs to kill cancer cells by attacking cells that are dividing quickly.2 However, other cells are also affected, such as those in the bone marrow, the lining of the mouth and intestines, and the hair follicles. This can lead to drug AEs. AEs depend on the type and dosage of drugs, how they are administered, and the length of therapy.2 Common AEs of systemic ChT include hair loss, mouth sores, loss of appetite, nausea and vomiting, diarrhea or constipation, increased risk of infections, bruising and/or bleeding, and fatigue.2 Some drugs may have other AEs. Drugs such as ondansetron, promethazine, and diphenhydramine may be used to control nausea and vomiting.
Untreated retinoblastoma is almost invariably fatal. With treatment, survival has increased from about 5% in 1930 to approximately 95% today.2,21 ChT is injected directly into the ophthalmic artery (the main artery supplying blood to the eye). Drugs often used in protocols or alone include vincristine, etoposide, and carboplatin (VEC) (TABLE 3),22 melphalan, and topotecan. This process may be repeated every few weeks, depending on how much the tumor shrinks.
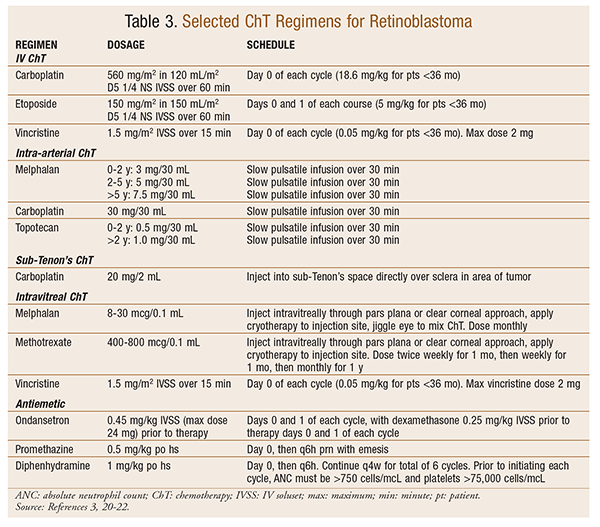
UMs usually respond poorly to standard ChT.1,2 ChT is used only when the cancer has become widespread. If ChT is used, the treatment is generally the same as for skin melanomas.
Immunotherapy: Immunotherapies, cytokines, monoclonal antibodies, and vaccines are among the most promising approaches for treating melanoma and lymphoma.2,23 These drugs stimulate the body’s immune system to attack cancer cells. Ipilimumab (Yervoy), a monoclonal antibody, boosts the overall activity of the immune system. Drugs such as nivolumab and pembrolizumab (Keytruda), which stimulate the immune system’s response against cancer cells, are being tested against skin melanomas. These drugs may prove useful against eye melanomas as well.
Targeted Therapy: Although targeted drugs and standard ChT drugs work differently, targeted therapy may work when ChT is ineffective.2,23 Most eye melanomas have changes in the GNAQ or GNA11 genes, and proteins made by these genes are part of the MAPK gene signaling pathway that helps cells grow. The following agents are not approved specifically for eye cancer, but they are being studied and used for the treatment of some eye cancers. Selumetinib targets the MEK protein, which is also part of the MAPK pathway. It has been shown to slow the growth of advanced eye melanomas; although it does not cure these cancers, it temporarily shrinks them. Sotrastaurin (AEB071), an investigational drug that targets protein kinase C, may be effective against cells with a GNAQ mutation. Drugs such as sunitinib (Sutent), sorafenib (Nexavar), vorinostat (Zolinza), and everolimus (Afinitor) are being used to treat certain types of cancer, and they could help treat eye cancers. Antiangiogenic drugs such as bevacizumab (Avastin) target blood vessels that tumors need to grow.
Pharmacist’s Role
It is unclear exactly what causes eye cancers, but there is generally believed to be a link between skin melanomas and sunlight exposure.1,2 Pharmacists are in a unique position to direct and educate patients and families about potential problems resulting from excessive exposure to direct sunlight and to suggest some basic strategies. Patients should limit exposure time to intense sunlight as much as possible; wearing a protective, wide-brimmed hat may also reduce the risk of eye melanomas. Eye cancers are uncommon, but all individuals should receive regular eye examinations by an ophthalmologist as a part of comprehensive health care. The American Cancer Society recommends wearing UV-protection sunglasses in strong sunlight, especially when a person is outside for extended periods.9 Wraparound sunglasses with 99% to 100% UVA and UVB absorption provides the best protection for the eyes and surrounding skin.
If a patient is concerned about a symptom or sign, medical attention should be sought. If ChT is appropriate, the pharmacist may consult with the patient’s oncologist on drug dosing, serve as a clinical monitor, and counsel the patient on the AEs of ChT and the importance of medication compliance. Hair loss, nausea and vomiting, and lethargy are fairly common AEs when IV ChT is used. The pharmacist can work with the patient and his or her oncologist to get prescription medication to manage these AEs and can reassure the patient that these AEs usually disappear or lessen in intensity after the treatment is finished. The pharmacist can encourage all patients to get routine eye examinations to check for changes in vision, as well as for eye cancers. The pharmacist may also assure the patient of his or her availability to answer questions about eye cancers, treatment, and appropriate sunscreen products.
REFERENCES
1. Eye cancer (melanoma and lymphoma). www.cancer.org/acs/groups/cid/documents/webcontent/003100-pdf.pdf. Accessed February 28, 2015.
2. Eagle RC Jr. The pathology of ocular cancer. Eye (Lond). 2013;27:128-136.
3. Retinoblastoma. http://medical-dictionary.thefreedictionary.com/retinoblastoma. Accessed May 5, 2015.
4. Verhoeff FH, Jackson E. Minutes of the proceedings of the 62nd annual meeting. Tran Am Ophthalmol Soc. 1926;24:38-43.
5. Gragoudas ES, Lane AM, Shih HA, et al. Uveal and conjunctival melanomas. UpToDate. www.uptodate.com/contents/uveal-and-conjunctival-melanomas. Accessed April 10, 2015.
6. Henriquez F, Janssen C, Kemp EG, Roberts F. The T1799A BRAF mutation is present in iris melanoma. Invest Ophthalmol Vis Sci. 2007:48;4897-4900.
7. Hocker T, Tsao H. Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Hum Mutat. 2007;28:578-588.
8. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410-1413.
9. Cancer.Net. Eye cancer: symptoms and signs. www.cancer.net/cancer-types/eye-cancer/symptoms-and-signs. Accessed May 26, 2015.
10. American Cancer Society. What are the key statistics for eye cancer? www.cancer.org/cancer/eyecancer/detailedguide/eye-cancer-key-statistics. Accessed February 28, 2015.
11. Shah CP, Weis E, Lajous M, et al. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112:1599-1607.
12. Schmidt-Pokrzywniak A, Jöckel KH, Bornfeld N, et al. Positive interaction between light iris color and ultraviolet radiation in relation to the risk of uveal melanoma: a case-control study. Ophthalmology. 2009;116:340-348.
13. Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397-2405.
14. Onken MD, Worley LA, Long MD, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008:49;5230-5234.
15. Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599-602.
16. Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010:363;2191-2199.
17. National Cancer Institute. Surveillance, Epidemiology, and End Results Program. http://seer.cancer.gov/statistics/summaries.html. Accessed February 28, 2015.
18. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010:17;1471-1474.
19. Cancer.Net. Eye cancer: stages and grades. www.cancer.net/cancer-types/eye-cancer/stages-and-grades. Accessed February 28, 2015.
20. American Cancer Society. How is retinoblastoma staged? www.cancer.org/cancer/retinoblastoma/detailedguide/retinoblastoma-staging. Accessed April 30, 2015.
21. Eagle RC Jr. Ocular tumors: triumphs, challenges and controversies. Saudi J Ophthalmol. 2013;27:129-132.
22. Shields CL, Fulco EM, Arias JD, et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye (Lond). 2013;27:253-264.
23. American Cancer Society. What’s new in eye cancer research and treatment? www.cancer.org/cancer/eyecancer/detailedguide/eye-cancer-new-research. Accessed March 1, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.



