US Pharm. 2016;41(11)(Specialty & Oncology suppl):7-12.
ABSTRACT: A biosimilar is a biological product designed to imitate a reference biologic with high similarities in structure, efficacy, and safety. Unlike generics, which are bioequivalent to the reference drug, most biosimilars are not approved as interchangeable products. The potential benefits of biosimilars are substantial, including immense savings in public health spending. As first-generation cancer biologics experience loss of patent protection, it can be expected that more biosimilars will be in the pipeline over the next several years. There are currently four FDA-approved biosimilars available in the United States, but only one (Zarxio) has cancer indications. Several other cancer biosimilars are also in the FDA approval process. There is an urgent need for clinicians to become increasingly aware of the regulation challenges and clinical impact of biosimilars.
Biologics are designed to treat diseases that may be rare or difficult to manage, such as cancer.1 Unlike synthetic small molecule generics, biologics are large molecules that are produced by living organisms.2 Biologics can include compounds such as blood products, vaccines, monoclonal antibodies, hormones, peptides, and growth factors (TABLE 1).2-10 While generics may be identical to their original pharmaceutical drugs, biosimilars are similar but not identical to their reference biologics.3,4 This is due to the fact that in the biosimilar development process, the developers do not have any knowledge of the processes used in the development of the original biological product.4,5
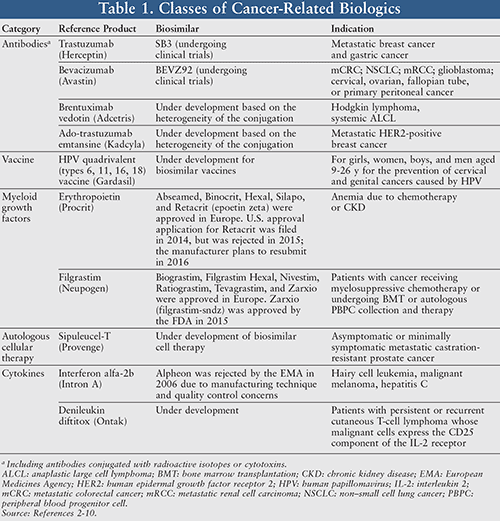
As many biologics are approaching loss of exclusivity, the focus has shifted to the development of biosimilars.2,3 It is therefore crucial to gain an understanding of the regulatory processes involved. Per the Biologics Price Competition and Innovation (BPCI) Act of 2009, a biosimilar must be “highly similar” to the reference biologic.1 The BPCI Act recognizes that biosimilars will not be identical to the original, as in the case of generic drugs; however, significant similarities should exist in terms of safety and efficacy.1 According to the FDA, there should be “no clinically meaningful differences between the biological product and the reference product for safety, purity, and potency of the product.”4
Terminology
Biologics are more complex than traditional chemical drugs, as they are created by or derived from living organisms. Development of biologics began in the 1980s as hormones and enzymes. Since then, the number of biologic categories has grown significantly.2 The terminology and approval process of biosimilars are complicated and may not be well understood. As drug development continues to bring novel biological products to the U.S. market, it is crucial to understand the difference between biologics, bio-similars, interchangeables, biomimics, and second-generation agents such as biobetters (TABLE 2).2,4,6,11-13
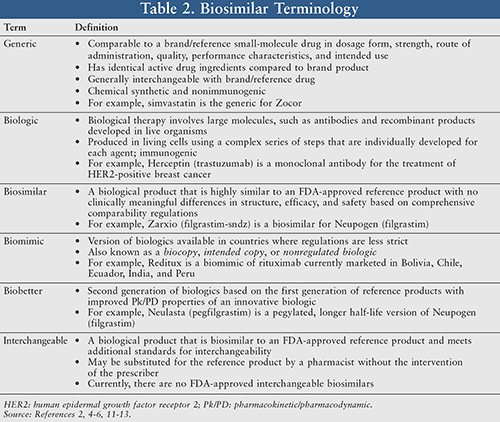
Biosimilars are not generic versions of the reference product—they are biologically unique. Biosimilars must be proven to be highly similar to the reference product in structure, efficacy, and safety through analytical, animal, and clinical studies in order to be approved by the FDA. The term biosimilar was first introduced in Europe in 2005 after idea development started in 2001, and was first used by the FDA in 2010.13
Unlike biosimilars, generics are considered interchangeable from their chemical reference product due to their proven identical structure and bioequivalence. A biosimilar is not considered interchangeable unless it is supported by multiple studies to demonstrate “the same clinical result” as the reference product and that switching between the biosimilar and the reference product carries no increased risk compared to using the reference product alone.4,12,14 The FDA regulations for interchangeable biologics are expected to be more stringent than those for biosimilars.2,11,13
The FDA has not yet published standards for interchangeability, but it plans to do so by the end of 2017.15 Though this means products cannot receive an “interchangeable” designation yet, Sandoz, for example, has conducted a clinical trial showing patient groups that alternated between Neupogen and its biosimilar Zarxio without differences in efficacy, safety, or anti-drug-antibody (ADA) development. This would appear to comply with the language of the BPCI Act, so it is possible Zarxio would obtain the designation soon after the standards are published.16
While biosimilars are similar to the reference product, biobetters are designed to exhibit enhanced efficacy or safety in comparison.6 Biobetters can have improved outcomes for the patient in terms of the formulation, dosing frequency, side-effect profile, or longer half-life. Essentially, biobetters are biologics that have been changed structurally for an improved outcome, whereas biosimilars mimic biologics. The clear distinction between biosimilars and biomimics is that biomimics are versions of biologics available in countries where regulation is less strict.6
FDA-Approved Biosimilars
As of October 2016, there are four FDA-approved biosimilars.17-20 Zarxio (filgrastim-sndz) had been the first and only FDA-approved biosimilar in the United States until Inflectra (infliximab-dyyb) was approved on April 5, 2016. Zarxio is the biosimilar to Neupogen (filgrastim), and it is currently the only FDA-approved biosimilar with an indication for cancer.17 Inflectra is the biosimilar for Remicade (infliximab), and it is approved for the treatment of multiple inflammatory diseases, including Crohn’s disease, ulcerative colitis, rheumatoid arthritis, and plaque psoriasis.18 Erelzi (etanercept-szzs), a biosimilar to Enbrel (etanercept), and Amjevita (adalimumab-atto), a biosimilar to Humira (adalimumab), were recently approved and are also indicated for multiple inflammatory and autoimmune disorders.19,20
Cancer Biosimilars
Neupogen is a granulocyte colony-stimulating factor (G-CSF) used in cancer patients with chemotherapy complications such as febrile neutropenia. Neupogen had recently lost its exclusivity, and in March 2015, Zarxio became the first biosimilar approved in the U.S. Another G-CSF, Granix (tbo-filgrastim), was approved by the FDA in 2012; however, because the type of application completed was a 351(a) instead of a 351(k) (discussed later), it cannot be considered a biosimilar nor a generic. It is worth contrasting these products with Neulasta (pegfilgrastim), filgrastim with an N-terminal PEGylation, which is a biobetter of Neupogen. Neulasta, approved as a 351(a) in 2002, cannot be considered biosimilar because not only is it not as similar as possible to Neupogen, but the modification actually improves on filgrastim by reducing renal clearance and prolonging in vivo persistence, which means there are significant clinical differences.21 TABLE 3 shows a comparison of the four FDA-approved G-CSF agents.22-26
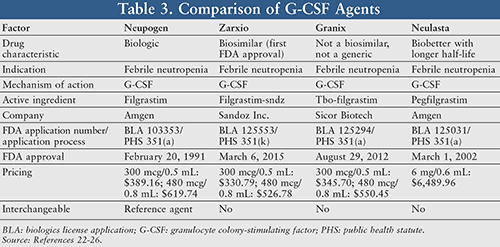
Zarxio is indicated for the treatment of patients with febrile neutropenia and in treating symptoms associated with chemotherapy in patients with acute myeloid leukemia, including fever and neutrophil recovery. In addition, Zarxio is indicated as a precursor for leukapheresis for use in hematopoietic stem cell transplantation (HSCT).23 When Zarxio was shown to be biosimilar to Neupogen and approved by the FDA, extensive tests were done on multiple levels before the conclusion was reached (TABLE 4).23 Biosimilarity between Zarxio and Neupogen was confirmed by the PIONEER study and a pharmacokinetic/pharmacodynamic (Pk/PD) study. Breast cancer patients with myelosuppressive chemotherapy (n = 218) were enrolled in a randomized, double-blind, phase III study with the primary endpoint of equivalency analysis for similar mean duration of severe neutropenia (DSN) between the Zarxio treatment group and the Neupogen group (mean DSN: Zarxio 1.17 days vs. Neupogen 1.2 days; 90% CI: -0.21 to 0.28; no significant difference). Since the manufacturer did not provide the evidence for interchangeable approval, Zarxio is not interchangeable with Neupogen.23 The safety profile of Zarxio as compared to Neupogen was not markedly different.16 Adverse effects are similar to the reference product and include nausea, fever pain, dyspnea, rash, fatigue, and thrombocytopenia.25
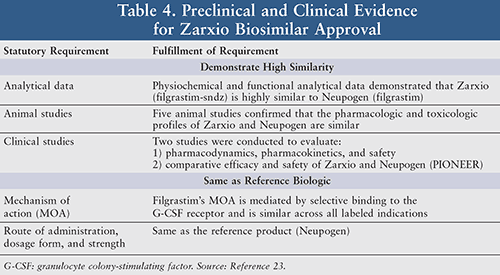
The efficacy of Granix was evaluated in a multinational, multicenter, randomized, controlled phase III study comparing Granix with placebo in 348 breast cancer patients treated with myelosuppressive chemotherapy. DSN was reduced by 71% in the Granix treatment group compared to the placebo group (mean DSN: Granix 1.1 days vs. placebo group 3.8 days; P <.0001).24
There are many other oncology biologics with biosimilars in development. The patent of pegfilgrastim (Neulasta) expired in 2015. The biologics license application (BLA) under the 310(K) pathway for CHS-1701, a biosimilar of Neulasta, was filed in August 2016 with supporting data of Pk/PD and immunogenicity studies. Rituximab (Rituxan) and cetuximab (Erbitux) are expiring in 2016. The patents of Herceptin (trastuzumab) and Avastin (bevacizumab) are expected to expire in 2019. The biosimilars STI-001 of Erbitux, CT P10 of Rituxan, SB3 of Herceptin, and BEVZ92 of Avastin are currently undergoing clinical trials.27-31
Benefits and Challenges of Biosimilars
Costs: Biologics are extremely expensive agents and comprise a significant portion of total health expenditures. The annual cost of a biologic treatment can reach $150,000.1 The market for biologics has been expanding significantly, especially in oncology treatments. For example, in 2010, over 55% of the money spent on cancer drugs at outpatient clinics was on biologics.1
In 2011, about one-fifth of the world’s pharmaceutical market was from the sales of monoclonal antibodies, which was valued at over $140 billion.1,32 Seven of the top 10 pharmaceutical products by global sales in 2014 were biologics. Three of the top-selling biologics are for cancer treatments, including Rituxan (ranked 6th with $7.36 billion); Avastin (7th with $6.84 billion); and Herceptin (9th with $6.69 billion). Cheaper biosimilars will generate tens of billions of dollars in savings for public health over the next decade. Most importantly, it may make treatment feasible for some patients who otherwise could not afford these therapies.1,32
There have been a number of predictions regarding how much less a biosimilar will cost compared to the reference product, which has varied between 12% and 51%, depending on the drug and predictor.33 Two notable bodies, the Congressional Budget Office and the RAND Corporation, estimated a 20% to 40% and 35% discount, respectively.33 That Zarxio costs a mere 15% less than Neupogen might appear to dash these expectations.34 Yet when there is a single generic for a small-molecule drug, the FDA found a mere 6% savings, which jumps to 48% when a second generic comes to market and 67% with five generics.35 Thus, it can be expected that the discount of biosimilars will markedly increase as more of them are approved for each reference biologic.33
Approval Process: Due to the complexity of biologics, the approval process for biosimilars has to be unique compared to that of generics. Small-molecule drugs are regulated by the Federal Food, Drug, and Cosmetic Act (FDCA) of 1938, which was later modified under the Hatch-Waxman Act of 1984.22 Biologics are regulated by the Public Health Services (PHS) Act of 1944. The FDA released guidelines about biosimilar approval in 2012 and 2015. These guidelines were created after the BPCI Act of 2009, which went into effect in 2010 as part of the Affordable Care Act.2 This act established an abbreviated licensure pathway for biosimilars. Three main stages constitute the process for biosimilar approval: 1) preclinical study with in vitro and in vivo data, 2) Pk/PD data in phase I clinical trials, and 3) phase III trials to confirm no clinical differences in safety and efficacy.13 The overarching challenge in the approval of biosimilars is balancing certainty of safety and efficacy with throughput of drugs.
Since the BPCI Act was passed, the FDA has designed a process that is in many ways more difficult and complex than that of some reputable foreign regulatory bodies. Biosimilars can go through sections 351(a) and 351(k) of the PHS Act for approval. Only drugs approved through 351(k) are considered biosimilar or interchangeable, which include a comprehensive report with analytical, preclinical, Pk/PD, and clinical data that demonstrate significant similarity between the biosimilar and the reference biologic, and comparable efficacy and safety.13,22 It is always beneficial to contact the appropriate FDA review division to define whether any of the analytical, preclinical, or clinical data can be bypassed in a 351(k) BLA.
Once approved, biosimilar products will be included in the FDA’s Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations, known as the Purple Book. This document lists all products approved under section 351 and several pieces of information for each, including whether they are biosimilar or interchangeable, and is analogous to the Orange Book. It is important to note that the Purple Book does not list all approved biologics, only those approved under section 351.36 Many biologics were approved under section 505 of the FDCA (e.g., insulins and somatropin products), though these 505 licenses will be transitioned to 351 licenses in 2020 as per the BPCI act, and presumably the products will then be listed in the Purple Book.37
Naming Biosimilars: Much controversy has taken place regarding the nonproprietary naming of biosimilars. On the one hand, if biosimilars share names with the reference product, then utilization, marketing, market penetration, and substitution would all be facilitated. On the other hand, unique naming would enable pharmacovigilance and tracking as well as help prevent unintended substitu-tions.38 Unsurprisingly, considering how different naming schema would affect biosimilar sales, Sandoz has come out in favor of shared names while Amgen calls for unique names.39,40
The FDA has taken the middle path in this case; biosimilars will have the same nonproprietary name as their reference product but with a four-letter suffix, “-sndz” in the case of Zarxio. This is meant to reap the benefits of both approaches—the shared portion will allow providers and patients to recognize the relationship of the biosimilar and reference product, while the suffix should prevent inadvertent substitution. The FDA has not yet decided whether or not interchangeable products, for which substitution is not potentially harmful, need to include a suffix in their name.41
The use of a suffix differs from the FDA history of using a prefix to differentiate but connect related biological products.42 Neulasta, the PEGylated form of filgrastim, is named pegfilgrastim.43 Teva was ordered to name Granix with the convention [xxx]-filgrastim, resulting in tbo-filgrastim.42 A prefix has also been added to the same product if it has different delivery and indication, as with Regeneron Pharmaceutical’s Eylea (aflibercept), an intraocular injection for wet macular degeneration, and Zaltrap (ziv-aflibercept), which has the same active biologic administered intravenously to treat colorectal cancer.43-45
Conclusion
As the loss of exclusivity for various biological products approaches, there are many opportunities for the pharma-ceutical industry to invest in the development of biosimilars. This requires clinicians to have a strong understanding of the drug development process for biosimilars, as well as the regulation challenges for their approval. Zarxio, the first and only FDA-approved cancer biosimilar available in the U.S., has opened the doors for an important field of research in biosimilars. Several cancer biosimilars, including SB3 for trastuzumab and BEVZ92 for bevacizumab, are currently undergoing clinical trials. With upcoming expiring patents for more biologics, additional research will be needed to overcome the challenges of developing and gaining approval for new biosimilars. The growing market for biosimilars is due to its advantages in providing wide access to patients in terms of affordability and availability.
REFERENCES
1. Singh SC, Bagnato KM. The economic implications of biosimilars. Am J Manag Care. 2015;21(16 suppl):s331-s340.
2. Epstein MS, Ehrenpreis, ED, Kulkarni PM. Biosimilars: the need, the challenge, the future. The FDA perspective. Am J Gastroenterol. 2014;109(12):1856-1859.
3. Zelenetz AD, Ahmed I, Braud EL, et al. NCCN biosimilars white paper: regulatory, scientific, and patient safety perspectives. J Natl Compr Canc Netw. 2011;9(suppl 4):S1-S22.
4. FDA. Information for healthcare professionals (biosimilars). www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241719.htm. Accessed May 1, 2016.
5. Stevenson JG. Clinical data and regulatory issues of biosimilar products. Am J Manag Care. 2015;21(16 suppl):s320-s330.
6. CDER therapeutic biologic products. Latest update March 2016. www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM164641.pdf. Accessed May 1, 2016.
7. Chen L, Wang L, Shion H, et al. In-depth structural characterization of Kadcyla (ado-trastuzumab emtansine) and its biosimilar candidate. MAbs. 2016;8(7):1210-1223.
8. Cohen A, Royzman I. FDA rejects Hospira’s EPO biosimilar application. The Biologics Law Blog of Patterson Belknap Webb & Tyler LLP. www.biosimilarscme.org/newsfeed/98-fda-rejects-hospira-s-epo-biosimilar-application. Accessed October 10, 2016.
9. Misra M. Biosimilars: current perspectives and future implications. Indian J Pharmacol. 2012;44(1):12-14.
10. Greer FM. Biosimilar development: from science to market. Life Sci Tech Bulletin. 2011;47:1-6.
11. Olech E. Biosimilars: rationale and current regulatory landscape. Semin Arthritis Rheum. 2016;45(5 suppl):S1-S10.
12. Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111-5117.
13. Dörner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11(12):713-724.
14. Ventola CL. Biosimilars—part 1: proposed regulatory criteria for FDA approval. P&T. 2013;38(5):270-287.
15. FDA. Biosimilar biological product reauthorization performance goals and procedures fiscal years 2018 through 2022. www.fda.gov/downloads/ForIndustry/UserFees/BiosimilarUserFeeActBsUFA/UCM521121.pdf. Accessed October 10, 2016.
16. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
17. FDA approves first biosimilar product Zarxio. FDA news release. March 6, 2016. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed October 10, 2016.
18. FDA approves Inflectra, a biosimilar to Remicade. FDA news release. April 5, 2016. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Accessed October 10, 2016.
19. FDA approves Erelzi, a biosimilar to Enbrel. FDA news release. August 30, 2016. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed October 10, 2016.
20. FDA approves Amjevita, a biosimilar to Humira. FDA news release. September 23, 2016. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed October 10, 2016.
21. Neulasta (pegfilgrastim) physician package insert. Thousand Oaks, CA: Amgen Inc; 2005. www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4185B1_03_09-FDA-Tab5b.pdf. Accessed October 10, 2016.
22. FDA. Information for industry (biosimilars). www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241720.htm. Accessed May 1, 2016.
23. Sandoz is pioneering the new FDA pathway: confirmed biosimilarity of Zarxio to Neupogen (filgrastim). Sandoz Inc. www.zarxio.com/info/hcp/pioneer.jsp?usertrack.filter_applied=true&NovaId=4029462171813837568. Accessed May 1, 2016.
24. GRANIX demonstrated a 71% reduction in duration of severe neutropenia (DSN) vs placebo. Cephalon, Inc. www.granixhcp.com/Clinical-information/GRANIX-efficacy.aspx. Accessed May 1, 2016.
25. Zarxio (filgrastim-sndz) package insert. Princeton, NJ: Sandoz Inc; March 2015. www.zarxio.com/assets/pdf/zarxio-pi.pdf. Accessed May 1, 2016.
26. FDA. Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research. Application number: 125031/0. Pegfilgrastim (Neulasta). January 31, 2002. www.accessdata.fda.gov/drugsatfda_docs/nda/2002/125031_ 000_Neulasta_medr_P1.pdf. Accessed October 10, 2016.
27. Pivot X, Curtit, E, Lee YJ, et al. A randomized phase I pharmacokinetic study comparing biosimilar candidate SB3 and trastuzumab in healthy male subjects. Clin Ther. 2016;38(7):1665-1673.
28. Clinical trials. Bioequivalence study bevacizumab biosimilar (BEVZ92) versus bevacizumab (AVASTIN) in first-line treatment mCRC patients. https://clinicaltrials.gov/ct2/show/NCT02069704. Accessed May 1, 2016.
29. Rugo HS, Linton KM, Cervi P, et al. A clinician’s guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
30. Generics and Biosimilars Initiative (GaBI Online). Biosimilars of cetuximab. April 1, 2016. http://gabionline.net/Biosimilars/General/Biosimilars-of-cetuximab. Accessed October 10, 2016
31. Coherus Biosciences. CHS-1701 pegfilgrastim biosimilar. www.coherus.com/our-products/chs-1701/. Accessed October 10, 2016.
32. Top 50 pharmaceutical products by global sales; 2014. PMLiVE. www.pmlive.com/top_pharma_list/Top_50_pharmaceutical_products_by_global_sales. Accessed May 1, 2016.
33. Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. RAND Corporation. 2016. www.rand.org/content/dam/rand/pubs/perspectives/PE100/PE127/RAND_PE127.pdf. October 10, 2016.
34. Raedler LA. Zarxio (filgrastim-sndz): first biosimilar approved in the United States. Am Health Drug Benefits. 2016,9:150-154.
35. Shapiro RJ, Singh K, Mukim M. The potential American market for generic biological treatments and the associated cost savings. Sonecon, LLC. February 2008. www.amcp.org/WorkArea/DownloadAsset.aspx?id=11651. Accessed October 10, 2016.
36. FDA. Background information: Lists of Licensed Biological Products With Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations (Purple Book). www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411424.htm. Accessed October 10, 2016.
37. FDA. Implementation of the “Deemed to be a License” provision of the Biologics Price Competition and Innovation Act of 2009. Guidance for Industry (draft guidance). March 2016. www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm490264.pdf. Accessed October 10, 2016.
38. Radar RA. Nomenclature of new biosimilars will be highly controversial. BioProcess Intern. June 1, 2011. www.bioprocessintl.com/manufacturing/biosimilars/nomenclature-of-new-biosimilars-will-be-highly-controversial-316512/. Accessed October 10, 2016.
39. Sandoz Inc. Sandoz position on the naming of biosimilars. www.sandoz-biosimilars.com/en/mediacenter/inn_naming_en.shtml. Accessed October 10, 2016.
40. Amgen. Amgen submits comments to FDA on biosimilars naming citizen petitions. December 20, 2013. www.amgen.com/media/our-perspective/archives/statements-and-responses/amgen-submits-comments-to-fda-on-biosimilars-naming-citizen-petitions/. Accessed October 10, 2016.
41. FDA. Nonproprietary naming of biological products. Guidance for industry (draft guidance). August 2015. www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm459987.pdf. Accessed October 10, 2016.
42. Center for Drug Evaluation and Research. Application number: 125294Orig1s00. Proprietary name review. BLA 125294 – [xxx]-filgrastim. August 2, 2012. www.accessdata.fda.gov/drugsatfda_docs/nda/2012/125294Orig1s000NameR.pdf. Accessed October 10, 2016.
43. FDA. Purple Book: List of Licensed Biological Products With Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. Accessed October 10, 2016.
44. FDA approves Eylea for eye disorder in older people. FDA news release. November 18, 2011. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm280601.htm. Accessed October 10, 2016.
45. National Cancer Institute. FDA approval for ziv-aflibercept. Updated July 3, 2013. www.cancer.gov/about-cancer/treatment/drugs/fda-ziv-aflibercept. Accessed October 10, 2016.
To comment on this article, contact rdavidson@uspharmacist.com.



