US Pharm. 2015;40(2):32-35.
ABSTRACT: Almost 6 million people in the United States have heart failure. When heart failure develops, cardiac output decreases and compensatory mechanisms activate. One of these mechanisms is cardiac fibrosis, a scarring process that over time impacts cardiac structure and function. Historically, cardiac fibrosis has not been a focus for treatment; however, it is now believed that therapy directed at cardiac fibrosis could reduce the progression of heart failure and other cardiovascular diseases. Medications that target the renin-angiotensin system, transforming growth factor-beta, and endothelin are in various stages of development.
Heart failure is a complex clinical syndrome in which structural or functional abnormalities impair the heart’s ability to fill with or pump blood.1,2 Affecting nearly 6 million people in the United States, heart failure is the leading reason for hospitalization in patients aged 65 years and older, as well as a major cause of impaired quality of life and chronic disability.1-3
Heart failure can result from systolic dysfunction, diastolic dysfunction, or both.1 The most common risk factors for developing heart failure include coronary heart disease (often with myocardial infarction [MI]), hypertension, diabetes, and cardiomyopathy.3,4
Heart failure is initiated by any event that impairs the heart’s ability to contract and/or relax, resulting in reduced cardiac output.1 As cardiac output decreases, compensatory mechanisms activate to restore cardiac output through increased preload, tachycardia, vasoconstriction, ventricular hypertrophy, and remodeling.1,4 At the cellular level, ventricular hypertrophy and remodeling are accompanied by cardiomyocyte hypertrophy, necrosis, apoptosis, fibroblast proliferation, and increased deposition of fibrous collagen, the last two of which are collectively termed cardiac fibrosis.4 This article will focus on cardiac fibrosis, including its mediators, assessment, and potential treatments.
Structure of the Heart Wall
The heart wall is composed of three layers: the epicardium (outer layer), the myocardium (middle layer), and the endocardium (interior layer).5 Fibrosis can occur in any layer and in various locations of the heart (i.e., the four heart chambers and valves).3 The discussion in this article will be limited to myocardial fibrosis.
The myocardium contains a variety of cell types, including cardiomyocytes and fibroblasts.6 Fibroblasts are found within the heart’s connective tissue and account for approximately two-thirds of cardiac cells.6-8 Fibroblasts are involved in many aspects of cardiac function, including regulating the balance of the extracellular matrix (ECM), ECM remodeling, electrical activity, production of growth factors and cytokines, and intercellular signaling.6
The structural component of the myocardium is the ECM.1 Collagen, a fibrous protein found in ECM and connective tissue, is composed of amino acids.1
Pathogenesis of Cardiac Fibrosis
Ventricular remodeling, a natural compensatory process that precedes the development of heart failure symptoms, results in progressive changes in the structure and function of the heart, including cardiac hypertrophy, loss of cardiac muscle cells, and ECM alterations.1 Some types of hypertrophy are accompanied by fibrosis.1
Cardiac fibrosis occurs when fibroblasts are activated to myofibroblasts and produce elevated amounts of ECM proteins that form scar tissue and alter normal degradation of ECM (FIGURE 1).4,7,8 Both processes lead to a buildup of collagen, which impacts both systolic and diastolic function.7,9
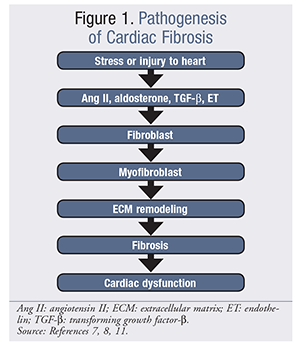
Cardiac fibrosis, which is part of the normal aging process, is associated with many cardiovascular diseases, including heart failure, hypertension, and cardiomyopathies; it also is found in hearts that have been damaged by MI or radiation.3,10-12 Fibrosis progresses over time and is accompanied by ongoing deterioration of heart function.13
Types of Fibrosis
Two forms of fibrosis—replacement fibrosis and reactive interstitial, or perivascular, fibrosis—have been identified.10 Replacement fibrosis occurs in response to an injury causing cardiomyocyte death, as in the case of MI; a reparative response is activated in the heart, causing replacement of dead cells and formation of a collagen-based scar.10,14 In reactive interstitial fibrosis, the cardiac interstitial space expands without significant cardiomyocyte loss.3,14 Reactive fibrosis allows the heart to adapt to injury and retain its pressure-generating ability.14 Pressure or volume overload, ischemia, and cardiomyopathies are examples of reactive fibrosis.14
Mediators of Fibrosis
Increases in various circulating hormones, cytokines, and proteins triggered by stress or injury contribute to fibroblast activation and differentiation and, ultimately, to cardiac fibrosis.7,8 Although many substances have been identified as playing a part in this process, several studies suggest that the renin-angiotensin system (RAS), transforming growth factor (TGF)-beta, and endothelin (ET) are key elements in the cascade. These elements will be the focus of this discussion.8
The RAS regulates the production and activity of fibroblasts.11 When the heart is injured, macrophages and fibroblasts produce renin and ACE, which in turn generate angiotensin II (Ang II).9 Ang II interacts with Ang II receptor type 1 (AT1) receptors, which promote hypertrophy, stimulate fibroblast proliferation, and increase collagen synthesis.9,15 In addition, Ang II suppresses collagenase (an enzyme that breaks down collagen), which may lead to increased collagen accumulation and fibrosis.13
Aldosterone has also been identified as a mediator of fibrosis. It affects fibroblast proliferation and collagen deposition in the ECM, heightening expression of cytokines and chemokines, signaling macrophages, and activating cardiomyocyte fibrogenic signals.1,9,12,13
TGF-beta is a cytokine in the heart that is activated by cardiac injury, generation of reactive oxygen species, Ang II, high glucose, altered pH, and certain proteases.4,9 Once activated, TGF-beta increases ECM production and decreases ECM breakdown.4,9
ET is a protein that is released by endothelial cells in the heart.8 It has been found to increase fibroblast differentiation into myofibroblasts and the production of ECM.8
Assessment of Fibrosis
Historically, the only measures of cardiac fibrosis have been echocardiograms that indirectly measure left ventricle mass and endomyocardial biopsies that measure collagen volume fraction (CVF).11 Newer techniques involve laboratory assessment of biomarkers and cardiac imaging.11
Several biomarkers for fibrosis have been identified, most of which are focused on ECM structure.11 Researchers can measure biomarkers of the synthesis and degradation of collagen types I and III, the main components of ECM.11 Collagen types I and III are synthesized as procollagens, which are then processed into mature collagen molecules by a peptidase cleaving their propeptide domain.6,11 During collagen synthesis, propeptides from the amino-terminals (PINP and PIIINP) or carboxy-terminals (PICP and PIIICP) of collagen types I and III are released and measured as biomarkers.6 During collagen degradation, telopeptides in the amino-terminals (NITP, NIIITP) or carboxy-terminals (CITP, CIIITP) of collagen types I and III are cleaved and act as biomarkers.6
Biomarkers may be used to identify fibrosis before symptoms of disease are present, as well as to assess the efficacy of medications. Biomarkers are commonly used as endpoints in clinical trials.11
MRI is useful for visualizing certain types of fibrosis.11 Contrast-enhanced cardiac MRI can readily visualize “patchy,” or regional, myocardial fibrosis from MI or infiltration.11,12 Diffuse fibrosis is much more difficult to visualize, and research into enhanced MRI techniques may prove beneficial in identifying this and other forms of fibrosis.11 Other imaging techniques that have shown promise in identifying cardiac fibrosis include single photon emission CT and positron emission tomography scanning; more studies are required, however.11
Potential Treatments
For certain conditions that cause fibrosis, such as volume or pressure overload, the best treatment is to prevent fibrosis by controlling risk factors.9,14 For patients in whom prevention is not possible or has failed, existing drugs and new chemical entities are in various stages of development; these products may have a significant effect on the management of many cardiovascular diseases.8,11
Agents That Impact the RAS: As stated previously, the RAS plays a central role in fibroblast activation; as such, it is an important target for drug therapy.11 A significant amount of research has been conducted on how drugs that modify this system affect cardiac fibrosis; specific information is provided in TABLE 1.
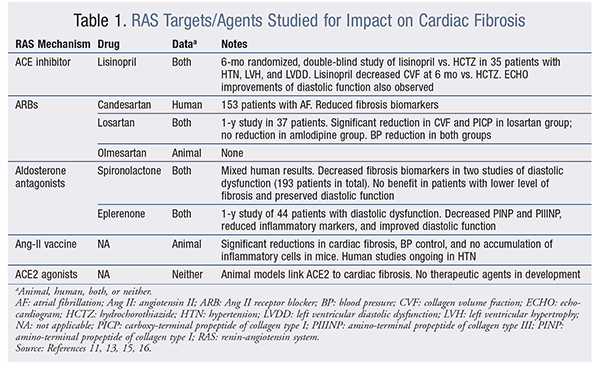
Several antihypertensive classes, including beta-blockers and calcium channel blockers, have shown efficacy in reducing fibrosis in animals; however, results in humans have been inconsistent.8,11 RAS inhibitors, such as ACE inhibitors, Ang II receptor blockers (ARBs), and aldosterone antagonists, have demonstrated positive results in animals and humans.11 One study of lisinopril versus hydrochlorothiazide (HCTZ) in patients with hypertension, left ventricular hypertrophy, and left ventricular diastolic dysfunction found that lisinopril decreased CVF significantly compared with HCTZ.13 Blood pressure (BP) was controlled in both treatment groups, but the effect on fibrosis differed.13
ARBs also are effective for reducing fibrosis.11 Losartan and olmesartan have favorable animal data, and candesartan and losartan have reduced fibrosis biomarkers in humans.8,11 In a 1-year study of losartan versus amlodipine, losartan significantly decreased fibrosis, whereas amlodipine did not; both drugs affected BP similarly, however.11 These studies of lisinopril and losartan show that treatments can impact fibrosis and hypertension independently of each other.11
Unlike ACE inhibitors and ARBs, aldosterone antagonists directly block aldosterone, which may be a more effective way of reducing its profibrotic effects.11 Spironolactone and eplerenone have been shown to reduce myocardial fibrosis in animals and humans, though results in humans are mixed.2,11
Vaccines with anti–Ang II effects are being researched in animals and humans, mostly for hypertension.11,16 One vaccine effectively decreased cardiac fibrosis in immunized mice; Ang II signaling was inhibited, and anti–Ang II antibodies increased.11
The RAS is a complex system with two counterbalancing axes.11,15 In addition to the familiar ACE/Ang II/AT1 axis, an ACE2/Ang-(1-7)/Mas receptor axis has been identified.11,15 ACE2 hydrolyzes Ang II into Ang-(1-7), and Mas is a protein receptor for Ang-(1-7).11,15 The ACE2/Ang-(1-7)/Mas receptor axis has shown antifibrogenic and antiproliferative effects in various organs, including the heart.15 Several animal studies have provided strong evidence that administration of Ang-(1-7) and overexpression of ACE2 can reduce cardiac fibrosis.15 While therapeutic agents for these areas have not yet been developed, the foundation for future research has been built.15
TGF-beta Inhibitors: As previously discussed, TGF-beta plays a central role in activating cardiac fibrosis, and inhibiting its actions could have profound effects on reducing fibrosis.4 Several approaches to inhibiting TGF-beta are being researched; two agents, pirfenidone and tranilast, are the furthest developed. Although the exact mechanisms of action are not completely understood, these drugs appear to inhibit TGF-beta and other growth factors.4
Pirfenidone, an oral medication, was approved in October 2014 for the treatment of idiopathic pulmonary fibrosis.17 In animal studies, pirfenidone has shown efficacy in reducing cardiac fibrosis, decreasing left atrial remodeling, and reducing diastolic stiffness, but not restoring cardiac contractility.4
Tranilast has been used in Japan for more than 20 years to treat asthma, allergic rhinitis, and atopic dermatitis.4,18 In animal studies, tranilast reduced cardiac fibrosis without affecting BP, suggesting a direct effect on fibrosis.4 In a human study examining prevention of restenosis after percutaneous coronary intervention, however, quantitative measures of efficacy were not found; moreover, several laboratory abnormalities were detected that could impact the use of tranilast, including increased bilirubin, liver enzymes exceeding three times the upper limit of normal, and serum creatinine increases of 50%. Research is being conducted on new compounds that could overcome some of these potential safety concerns.4
ET Inhibitors: Currently, several ET receptor inhibitors are approved in the U.S. for the treatment of pulmonary hypertension, including the dual ET subtype A/ET subtype B (ETA/ETB) inhibitors bosentan and macitentan and the ETA inhibitor ambrisentan.8,19-21 It is postulated that it may be necessary to target dual ETA/ETB inhibition, since both receptors impact fibrosis.22 In animal studies, bosentan has been demonstrated to inhibit ECM formation, decrease collagen synthesis, and increase collagenase suppression.22 Studies examining its use in fibrosis are ongoing.8
Drugs Impacting Other Mediators of Fibrosis: Histone deacetylases (HDACs) are enzymes that play a key role in regulating gene transcription throughout the body.7 HDACs may be linked to the signaling of some cellular molecules that have an effect on cardiac fibrosis, as well as inflammation.7 Animal studies have revealed that HDAC inhibitors can stop, or even reverse, cardiac fibrosis.7 One inhibitor has positive results in reducing cardiac fibrosis, as well as levels of Ang II receptors and TGF-beta.7 Although a great deal of research is needed, HDAC inhibitors hold promise as future treatment.7
Ivabradine is an oral medication currently available outside the U.S. that provides selective heart rate reduction by inhibiting the f-channel of the sinoatrial node.23 In August 2014, the FDA granted fast-track designation for this compound in the treatment of chronic heart failure.23 In animal models, ivabradine effectively reduced fibrosis and circulating Ang II and aldosterone levels.11
Additional agents, including diltiazem, tadalafil, isosorbide dinitrate and hydralazine, erythropoietin, cyclosporine, thalidomide, and anti-inflammatory drugs impacting cytokines (e.g., tumor necrosis factor-alpha, interleukin [IL]-1, and IL-6), are being evaluated for fibrosis.11 Transplantation of a variety of stem cells following MI has been demonstrated to decrease cardiac fibrosis and cardiac muscle apoptosis.11,14
Conclusion
Cardiac fibrosis is believed to be the final pathway leading to heart failure, which is an extremely common syndrome in the U.S. Much of the pathophysiology of cardiac fibrosis is known. Many drug treatments have been studied and seem promising, but data in humans are both limited and mixed. Although more studies must be conducted to evaluate the safety and efficacy of possible treatments for cardiac fibrosis, there is great hope for the future.
REFERENCES
1. Parker RB, Nappi JM, Cavallari LH. Chronic heart failure. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw-Hill Education; 2014.
2. Rich MW. Heart failure. In: Halter JB, Ouslander JG, Tinetti ME, et al, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. McGraw-Hill; 2009.
3. Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2:158-173.
4. Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-beta. Cardiovasc Ther. 2012;30:e30-e40.
5. Mescher AL. Junqueira’s Basic Histology: Text and Atlas. 13th ed. New York, NY: McGraw-Hill; 2013:212-233.
6. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15.
7. Tao H, Shi KH, Yang JJ, et al. Histone deacetylases in cardiac fibrosis: current perspectives for therapy. Cell Signal. 2014;26:521-527.
8. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675-1680.
9. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549-574.
10. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631-637.
11. Roubille F, Busseuil D, Merlet N, et al. Investigational drugs targeting cardiac fibrosis. Expert Rev Cardiovasc Ther. 2014;12:111-125.
12. Bashore TM, Granger CB, Jackson K, Patel MR. Heart disease. In: Papadakis MA, McPhee SJ, eds. Current Medical Diagnosis & Treatment 2015. New York, NY: McGraw-Hill Education; 2015.
13. Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388-1393.
14. Elnakish MT, Kuppusamy P, Khan M. Stem cell transplantation as a therapy for cardiac fibrosis. J Pathol. 2013;229:347-354.
15. Simões e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169:477-492.
16. Bairwa M, Pilania M, Gupta V, Yadav K. Hypertension vaccine may be a boon to millions in developing world. Hum Vaccin Immunother. 2014;30:708-713.
17. FDA. FDA approves Esbriet to treat idiopathic pulmonary fibrosis. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm418991.htm. Accessed October 27, 2014.
18. Kissei Pharmaceutical Co. Kissei entered into an agreement with Nuon Therapeutics, Inc on Tranilast. www.kissei.co.jp/e_contents/relation/2007/20070723.html. Accessed October 22, 2014.
19. Tracleer (bosentan) product information. South San Francisco, CA: Actelion Pharmaceuticals US, Inc; October 2012.
20. Letairis (ambrisentan) product information. Foster City, CA: Gilead Sciences, Inc; May 2014.
21. Opsumit (macitentan) product information. South San Francisco, CA: Actelion Pharmaceuticals US, Inc; October 2013.
22. Clozel M, Salloukh H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann Med. 2005;37:2-12.
23. Amgen. FDA grants Amgen priority review designation for ivabradine for the treatment of chronic heart failure. www.amgen.com/media/media_pr_detail.jsp?releaseID=1961392. Accessed October 22, 2014.
To comment on this article, contact rdavidson@uspharmacist.com.


