ABSTRACT: Pulmonary arterial hypertension (PAH) is a chronic condition that carries a significant risk of morbidity and mortality. PAH is generally thought to result from an imbalance of vasoconstrictive and vasodilatory mediators. The treatment of PAH includes calcium channel blockers, endothelin receptor antagonists, phosphodiesterase inhibitors, and prostacyclin analogues. New and emerging therapies include medications with novel molecular targets and dosing mechanisms. Pharmacists can play an integral role in managing the needs of patients with this serious condition.
Pulmonary arterial hypertension (PAH), a serious chronic condition, is a subcategory of pulmonary hypertension (PH). PAH is defined by a mean pulmonary arterial pressure (mPAP) greater than 25 mmHg at rest, pulmonary capillary wedge pressure (PCWP) less than 15 mmHg, and normal cardiac output.1-3 The prevalence and incidence of PAH are approximately 15 cases per million and 2.4 cases per million adult population per year, respectively.4,5 Medications known to induce PAH include anorexigenics (e.g., aminorex, fenfluramine, dexfenfluramine), amphetamines, toxic rapeseed oil, and serotonergic agents (selective serotonin reuptake inhibitors, St. John’s wort, pergolide).3 PAH is described extensively in the literature; therefore, most pharmacologic treatments target this subclassification of PH. Treatment centers on improving quality of life and functional status (e.g., 6-minute walk distance [6MWD]). The World Health Organization and New York Heart Association (NYHA) classification scales are often used interchangeably in practice to categorize functional status in PAH patients.3,6,7 The NYHA classification—in which class I denotes no limitations on activity and class IV denotes symptoms at rest—is used in this review.6,7
PATHOPHYSIOLOGY
The development of PAH is a multifactorial process involving an increase in pulmonary vascular resistance (PVR) due to vasoconstriction, proliferative and obstructive remodeling of pulmonary structures, inflammation, and thrombosis. Endothelial dysfunction leads to impaired production of vasodilators (e.g., nitric oxide [NO], prostacyclins, vasoactive intestinal peptides) and overexpression of vasoconstrictive and proliferative substances (e.g., thromboxane A2, endothelin [ET]-1). These alterations in physiobiological substances lead to permanent changes in lung parenchyma. The consequent increases in PVR lead to right ventricular (RV) overload, hypertrophy, dilation, RV failure, and death.3 All therapies, whether conventional or investigational, seek to restore the balance between the vasodilatory and vasoconstrictive processes in the lung.
SCREENING
Patients with known bone morphogenetic protein receptor type 2 mutation, portal hypertension, connective-tissue disorders, or HIV should be screened annually.8-10 Since 10% of PAH cases are inherited, patients with multiple affected family members should be screened.8-10 Genetic screening is not recommended, since only 10% to 20% of carriers develop the disorder.9 Less frequent screenings are warranted in cases of atrial septal defect or exposure to toxic agents.9
CLINICAL PRESENTATION
The clinical presentation of PAH includes slowly progressive dyspnea, fatigue, angina, and syncope. Lower-extremity edema, palpitations, and exercise limitation occur in advanced disease.9,10 Typically, diagnosis occurs 2 years after symptom onset and at a mean age of 37 years.9,10
Diagnosis involves several tests, including chest x-ray, electrocardiogram, and echocardiogram. Right heart catheterization to measure mPAP, PCWP, and left ventricular (LV) end-diastolic pressure and vasodilator testing should be performed.8-10 Vasodilator testing is conducted to determine whether the patient would respond to calcium channel blocker (CCB) therapy for primary symptom management.2
PHARMACOLOGIC TREATMENT OPTIONS
Conventional Therapies
CCBs: These agents cause smooth-muscle relaxation in the pulmonary and systemic vascular beds and result in vasodilation. High-dose CCB therapy improves NYHA functional class and extends survival.10 In two small cohort studies, survival rates at 5 and 7 years were 95% and 97%, respectively.11,12 Long-acting nifedipine, sustained-release diltiazem, and amlodipine are used clinically (TABLE 1).13 If the response to therapy is inadequate after 3 to 6 months, or if the patient is nonresponsive to vasoreactivity testing, therapy with prostacyclin analogues, endothelin receptor antagonists (ERAs), and/or phosphodiesterase type 5 (PDE-5) inhibitors should be initiated.8,14
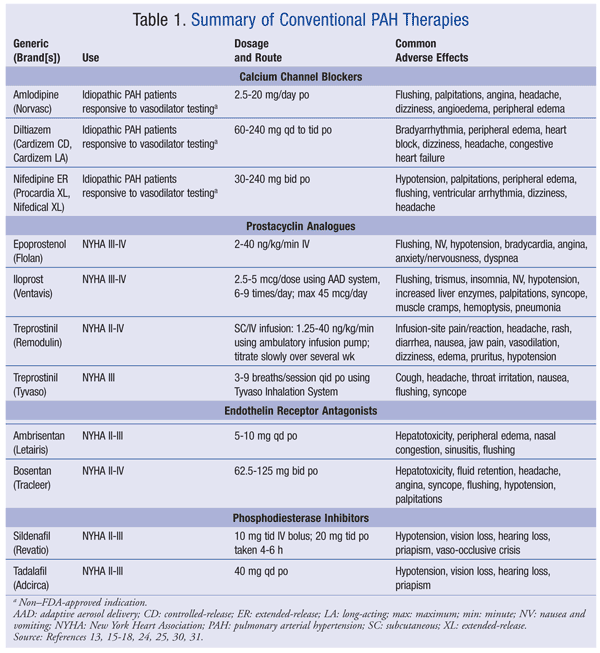
Prostacyclin Analogues: Three prostacyclin analogues are available in the United States: epoprostenol (Flolan), treprostinil (Remodulin, Tyvaso), and iloprost (Ventavis) (TABLE 1).15-18 Prostacyclin analogues cause direct vasodilation of the pulmonary and systemic arterial vascular beds and inhibit platelet aggregation.13 These agents may potentiate the hemodynamic effects of antihypertensives and the hematologic effects of anticoagulants and antiplatelets. Generally, they are available only through specialty pharmacies.
Epoprostenol improves functional class, exercise tolerance, hemodynamics, and survival.19,20 This agent should be administered via ambulatory infusion pump through central intravenous (IV) access. It should be initiated at 2 ng/kg/min and titrated at intervals of at least 15 minutes to the desired clinical effect. Abrupt discontinuation may lead to pronounced rebound PH; therefore, to avoid interruptions in drug delivery, patients should have access to a backup infusion pump and IV infusion sets. Epoprostenol is contraindicated in patients who have chronic heart failure with severe LV dysfunction and in patients who develop pulmonary edema during treatment initiation. Reconstituted unused solution should be refrigerated and protected from light and may be kept for no more than 48 hours. Reconstituted epoprostenol may be administered at 25°C for 8 hours. A cold pouch may be used to extend the duration of administration for up to 24 hours.15
Treprostinil is available in subcutaneous (SC)/IV (Remodulin) and orally inhaled (Tyvaso) formulations, all of which lessen clinical deterioration, improve 6MWD, and confer survival benefits.16,17,21,22 Treprostinil may be administered through central IV access in patients who cannot tolerate SC infusion. The infusion is initiated at 1.25 ng/kg/min. The initial dose should be reduced by 50% in patients who cannot tolerate full doses or in those with hepatic impairment. The dosage should be titrated by 1.25 ng/kg/min at weekly intervals for the first month, then 2.5 ng/kg/min weekly thereafter. When administered via SC infusion, treprostinil may be delivered without further dilution; however, it must be diluted when administered IV. Diluted treprostinil is stable for up to 48 hours at 25°C in concentrations as low as 4,000 ng/mL. In patients requiring transition from epoprostenol, treprostinil is generally initiated at 10% of the calculated dosage and gradually titrated while the epoprostenol is gradually tapered.
Serious adverse effects, such as infusion-site pain/reactions and sepsis, are associated with the systemic delivery method. There have been postmarketing reports of thrombophlebitis associated with peripheral IV infusion, thrombocytopenia, bone pain, and generalized rashes. Advantages of treprostinil are its standard dose-titration schedule and multiple methods of administration. Treprostinil may be initiated in mild-to-moderate disease and in heart failure.16
Orally inhaled treprostinil should be administered during waking hours via the manufacturer-supplied delivery system. It should be initiated at three breaths (18 mcg) per treatment, with a daily maximum of four treatments performed at least 4 hours apart. The dosage should be increased, in accordance with patient tolerance, by three breaths at 1- to 2-week intervals and titrated to a target of nine breaths (54 mcg) per treatment. Inhaled treprostinil has not been studied in patients with asthma or chronic obstructive pulmonary disease; therefore, its use in these patients may not be advisable. The bioavailability of inhaled treprostinil at the 18-mcg dosage is 64%; therefore, the same systemic adverse effects that are possible with this class of drugs should be expected with this agent. Inhaled treprostinil does not require an ambulatory infusion pump, so the risk of infection is reduced. One drawback is that the frequent dosing may impact adherence.17
Iloprost is an inhaled prostacyclin analogue that is administered via the corresponding inhalation delivery system. The dosage should be initiated at 2.5 mcg, then increased to 5 mcg if tolerated, and administered six to nine times per day (about every 2 hours). Dosing intervals should be extended in patients with hepatic impairment. Iloprost should be avoided in severe hypotension because of the increased risk of syncope. Patients with pulmonary venous hypertension and bronchospasms and those with a history of airway hyperreactivity should not be given iloprost because of the risk of severe pulmonary edema in these patients.18 Iloprost improves functional class, 6MWD, and clinical symptoms in patients with PAH.23 This formulation offers the same advantages and disadvantages as inhaled treprostinil; however, iloprost is indicated for PAH patients with NYHA class III or IV symptoms, as opposed to orally inhaled treprostinil, which is approved only for NYHA class III PAH.
ERAs: Two nonselective ERAs are available in the U.S.: bosentan (Tracleer) and ambrisentan (Letairis) (TABLE 1).24,25 ERAs cause vasodilation by blocking the binding of ET-1 hormone to the ETA and ETB receptors. ETA receptors act primarily to mediate vasoconstriction and cell proliferation, whereas ETB receptors mediate vasodilation and ET-1 clearance. Although bosentan and ambrisentan affect both receptor subtypes, they have higher affinity for the ETA receptor, which has a net effect on vasodilaton.13 Data from 11 randomized or quasi-randomized trials in 1,457 patients indicate that ERAs improve dyspnea, functional class, and exercise capacity in patients with PAH.26 Both agents are known hepatotoxins; therefore, dose reduction with frequent monitoring or drug discontinuation should be considered if there is evidence of acute liver injury.24,25 Owing to the hepatotoxicity and teratogenicity of these agents, patients and health care professionals must enroll in monitoring programs and comply with the FDA’s Risk Evaluation and Mitigation Strategy for that drug (bosentan, Tracleer Access Program; ambrisentan, Letairis Education and Access Program). The patient must provide monthly pregnancy test results to the prescribing physician.24,25
Bosentan is indicated to improve exercise ability and delay clinical worsening.24 In randomized trials, bosentan has improved exercise tolerance, dyspnea, and functional class and has increased the time to clinical worsening.27,28 It also has been shown to improve cardiac index, PVR, and mPAP.28 Bosentan should be initiated at 62.5 mg orally twice daily and titrated to 125 mg twice daily after 4 weeks. Patients with a low body weight (<40 kg) should be maintained at the lower dosage. The dosing frequency should be reduced when bosentan is given in combination with ritonavir, with the bosentan being discontinued 36 hours before ritonavir initiation. Other major drug interactions involve oral contraceptives, simvastatin, and rifampin.24
Ambrisentan is indicated to improve exercise ability and delay clinical worsening. It should be initiated at 5 mg orally once daily, then titrated to 10 mg daily.25 Ambrisentan was shown to improve 6MWD, health-functioning scales, and dyspnea scores.29 It should not be used in combination with cyclosporine. Ambrisentan offers the advantage of once-daily dosing, which may improve compliance.25
PDE-5 Inhibitors: Two PDE-5 inhibitors are indicated for use in PAH: sildenafil (Revatio) and tadalafil (Adcirca) (TABLE 1).30,31 Phosphodiesterases are responsible for the degradation of cyclic guanosine monophosphate (cGMP)—a key mediator of vasodilation—in the smooth muscle. The PDE-5 receptor subtype is found predominantly in the pulmonary vasculature. PDE-5 inhibitors therefore increase the concentration of cGMP, resulting in relaxation of pulmonary vascular smooth-muscle cells and vasodilation of the pulmonary vascular bed. Sildenafil and tadalafil have additive blood pressure–lowering effects and should be not be used in patients taking nitrates, alpha blockers, or alcohol. Hearing loss, visual impairment, and priapism may occur with use of these PDE-5 inhibitors. Concomitant use of potent inducers and inhibitors of CYP450 3A should be avoided.30,31
Sildenafil is indicated to improve exercise ability and delay clinical worsening. In randomized short-term and long-term studies, sildenafil significantly improved exercise capacity, 6MWD, functional class, PAP, and PVR.32-34 Sildenafil is available as an oral tablet and suspension and an IV bolus injection. The oral tablet and suspension are dosed at 20 mg three times daily, and the injection is dosed at 10 mg IV bolus three times daily. There have been limited clinical trials of the combination of sildenafil and bosentan.30
Tadalafil is approved to improve exercise capacity in PAH patients. One randomized trial demonstrated increased 6MWD and time to clinical worsening. Of note, 50% of patients were also receiving bosentan.31 Over 16 weeks, tadalafil produced a mean 44-meter increase in 6MWD.35 Tadalafil, which is available as a 20-mg tablet, is dosed at 40 mg once daily. It should be dosed at 20 mg daily in mild-to-moderate renal and hepatic disease and should be avoided in severe disease. Once-daily dosing may improve compliance; however, caution must be taken in special populations.31
Investigational Therapies
Research is currently under way to evaluate the utility of multiple new molecular targets for PAH treatment. Fasudil, a Rho-kinase inhibitor currently in phase I trials, inactivates myosin light chain (MLC) phosphatase, thereby shifting MLC equilibrium toward an unphosphorylated state resulting in vasorelaxation.36 Fasudil has a short half-life (45 minutes) and lacks selectivity for the pulmonary circulation.37,38
Riociguat, the first soluble guanylate cyclase (sGC) stimulator is currently in phase II trials. It works by mimicking the effects of NO and increasing the sensitivity of sGC to endogenous NO, thereby reducing the workload of the right ventricle.39,40
Imatinib is a platelet-derived growth factor receptor currently used for certain cancers. In phase III studies, it appears promising for the treatment of PAH.41 Imatinib has been shown to reduce PVR and improve 6MWD by lessening vascular smooth-cell proliferation in the pulmonary artery.41
Selexipag (oral selective prostacyclin receptor antagonist) and oral treprostinil (prostacyclin analogue) are undergoing phase II and III trials, respectively. Both agents have been shown to reduce PVR and systemic vascular resistance while increasing CI, and they may prove to be therapeutic alternatives to current prostacyclin analogues.42-44
Macitentan, a dual ERA currently in phase I trials, antagonizes the action of ET-1 on cell membranes. The benefit of using a dual ERA is that it reduces the crosstalk between receptors that causes increased expression of one subtype when the other subtype is blocked.45,46
GUIDELINE RECOMMENDATIONS
Guidelines developed in 2009 by the American College of Cardiology Foundation/American Heart Association (ACCF/AHA) recommend that appropriate general and supportive care measures, including education and anticoagulation, be provided to patients with PH. See TABLE 2 for a summary.

THE PHARMACIST’S ROLE
A medication history is important, since agents used to treat PAH may be dispensed by specialty pharmacies, whereas other agents are dispensed by community pharmacies. Counseling includes administration techniques.8 Monitoring includes medication safety, signs and symptoms, and quality of life, with physician visits every 3 to 6 months.18 Pneumococcal and influenza immunizations are recommended.8,9
Nondrug therapies include limiting sodium intake and aerobic exercise, as well as avoiding pregnancy, high altitudes, and heavy physical exertion.8,9 Patients may require oxygen when traveling on commercial aircraft.8
There is no consensus regarding the use of oral contraceptives. Options include low-dose estrogen with warfarin, surgical sterilization, and barrier methods.8,9 OTC vasoconstrictive medications should be avoided, and herbal products should be used with caution.47
REFERENCES
1. Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S43-S54.
2. Galiè N, Torbicki A, Barst R, et al. Guidelines on diagnosis and
treatment of pulmonary arterial hypertension. The Task Force on
Diagnosis and Treatment of Pulmonary Arterial Hypertension of the
European Society of Cardiology. Eur Heart J. 2004;25:2243-2278.
3. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis
and treatment of pulmonary hypertension: The Task Force for the
Diagnosis and Treatment of Pulmonary Hypertension of the European
Society of Cardiology (ESC) and the European Respiratory (ERS), endorsed
by the International Society of Heart and Lung Transplantation (ISHLT).
Eur Heart J. 2009;30:2493-2537.
4. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023-1030.
5. Peacock AJ, Murphy NF, McMurray JJ, et al. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104-109.
6. Rubin LJ; American College of Chest Physicians. Diagnosis and
management of pulmonary arterial hypertension: ACCP evidence-based
clinical practice guidelines. Chest. 2004;126(suppl 1):S7-S10.
7. Rubin LJ; American College of Chest Physicians. Diagnosis and
management of pulmonary arterial hypertension: ACCP evidence-based
clinical practice guidelines. Chest. 2004;126(suppl 1):S4-S6.
8. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert
consensus document on pulmonary hypertension: a report of the American
College of Cardiology Foundation Task Force on Expert Consensus
Documents and the American Heart Association: developed in collaboration
with the American College of Chest Physicians, American Thoracic
Society, Inc, and the Pulmonary Hypertension Association. Circulation. 2009;119:2250-2294.
9. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417-1431.
10. Edelman JD. Clinical presentation, differential diagnosis, and vasodilator testing of pulmonary hypertension. Semin Cardiothorac Vasc Anesth. 2007;11:110-118.
11. Rich S, Kaufmann E, Levy PS. The effect of high doses of
calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76-81.
12. Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium
channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105-3111.
13. Newman JH. Treatment of primary pulmonary hypertension—the next generation. N Engl J Med. 2002;346:933-935.
14. Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S78-S84.
15. Flolan (epoprostenol) product information. Research Triangle Park, NC: GlaxoSmithKline; March 2011.
16. Tyvaso (treprostinil) product information. Research Triangle Park, NC: United Therapeutics Corp; July 2012.
17. Remodulin (treprostinil) product information. Research Triangle Park, NC: United Therapeutics Corp; February 2011.
18. Ventavis (iloprost) product information. South San Francisco, CA: Actelion Pharmaceuticals US, Inc; August 2012.
19. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous
intravenous epoprostenol (prostacyclin) with conventional therapy for
primary pulmonary hypertension. N Engl J Med. 1996;334:296-301.
20. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477-1482.
21. Rubenfire M, McLaughlin VV, Allen RP, et al. Transition from IV
epoprostenol to subcutaneous treprostinil in pulmonary arterial
hypertension: a controlled trial. Chest. 2007;132:757-763.
22. Barst RJ, Galiè N, Naeije R, et al. Long-term outcome in
pulmonary arterial hypertension patients treated with subcutaneous
treprostinil. Eur Respir J. 2006;28:1195-1203.
23. Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322-329.
24. Tracleer (bosentan) product information. South San Francisco, CA: Actelion Pharmaceuticals US, Inc; October 2010.
25. Letairis (ambrisentan) product information. Foster City, CA: Gilead Sciences, Inc; October 2012.
26. Liu C, Chen J, Gao Y, et al. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst Rev. 2009;(3):CD004434.
27. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896-903.
28. Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual
endothelin-receptor antagonist bosentan in patients with pulmonary
hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119-1123.
29. Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the
treatment of pulmonary arterial hypertension: results of the ambrisentan
in pulmonary arterial hypertension, randomized, double-blind,
placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010-3019.
30. Revatio (sildenafil) product information. New York, NY: Pfizer; August 2012.
31. Adcirca (tadalafil) product information. Indianapolis, IN: Eli Lilly and Co; March 2012.
32. Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148-2157.
33. Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of
sildenafil in primary pulmonary hypertension: a randomized,
placebo-controlled, double-blind, crossover study. J Am Coll Cardiol. 2004;43:1149-1153.
34. Pepke-Zaba J, Gilbert C, Collings L, Brown MC. Sildenafil
improves health-related quality of life in patients with pulmonary
arterial hypertension. Chest. 2008;133:183-189.
35. Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894-2903.
36. Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol. 2008;155:444-454.
37. Fukumoto Y, Matoba T, Ito A, et al. Acute vasodilator effects of a
Rho-kinase inhibitor, fasudil, in patients with severe pulmonary
hypertension. Heart. 2005;91:391-392.
38. Ishikura K, Yamada N, Ito M, et al. Beneficial acute effects of
rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J. 2006;70:174-178.
39. Frey R, Mück W, Unger S, et al. Single-dose pharmacokinetics,
pharmacodynamics, tolerability, and safety of the soluble guanylate
cyclase stimulator BAY 63-2521: an ascending-dose study in healthy male
volunteers. J Clin Pharmacol. 2008;48:926-934.
40. Ghofrani HA, Hoeper MM, Halank M, et al. Riociguat for chronic
thromboembolic pulmonary hypertension and pulmonary arterial
hypertension: a phase II study. Eur Respir J. 2010;36:792-799.
41. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as
add-on therapy for pulmonary arterial hypertension: results of the
randomized IMPRES study. Circulation. 2013;127:1128-1138.
42. Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral,
selective prostacyclin receptor agonist for the treatment of pulmonary
arterial hypertension. Eur Respir J. 2012;40:874-880.
43. Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the
treatment of pulmonary arterial hypertension in patients on background
endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor
therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142:1383-1390.
44. Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral
treprostinil monotherapy for the treatment of pulmonary arterial
hypertension: a randomized, controlled trial. Circulation. 2013;127:624-633.
45. Iglarz M, Binkert C, Morrison K, et al. Pharmacology of
macitentan, an orally active tissue-targeting dual endothelin receptor
antagonist. J Pharmacol Exp Ther. 2008;327:736-745.
46. Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J.
Macitentan: entry-into-humans study with a new endothelin receptor
antagonist. Eur J Clin Pharmacol. 2011;67:977-984.
47. Pulmonary Hypertension Association. Consensus statement:
recommendations on over-the-counter medications in patients with PAH.
www.phaonlineuniv.org/DiagnosisTreatment/content.cfm?ItemNumber=1519.
Accessed March 13, 2013.
To comment on this article, contact rdavidson@uspharmacist.com.



