US Pharm. 2016;41(1):22-26.
ABSTRACT: Multiple sclerosis (MS) is an inflammatory disease of demyelinated axons and central nervous system (CNS) plaque formation. MS affects younger adults, occurs more frequently in wo men than in men, and may involve genetic, environmental, and immunologic factors. MS manifests as neurologic signs and symptoms related to declining neuromuscular, sensory, and cognitive function, with a waxing and waning pattern of symptoms and/or continuous disease progression. Diagnosis is based on clinical symptoms and MRI confirmation of the timing and location of CNS lesions. Drug treatment focuses on the immunologic effects of MS in an attempt to modify or halt disease progression and to manage associated clinical symptoms.
Multiple sclerosis (MS) is an immunologic disorder marked by chronic inflammation of the central nervous system (CNS).1 In MS, mononuclear cells infiltrate the perivascular space between arteries and veins and the pia mater, axons are demyelinated and destroyed, and immunoreactive changes in glial cells result in the formation of plaques in multiple areas of the CNS. Key to the diagnosis of MS is the dissemination of these plaques in time and space.1
Epidemiology
The onset of MS occurs most often between ages 20 and 50 years (mean 30 years), with a female-to-male ratio approximating 3-to-1.1 Although the cause of MS is unknown, various genetic, environmental, and immunologic factors have been implicated.1 An estimated 2 million to 2.5 million people worldwide have MS. Prevalence follows an unevenly distributed latitudinal gradient, with fewer than five in 100,000 cases in tropical areas or Asia and more than 100 to 200 in 100,000 cases in temperate regions with large populations of Northern European origin, including North America, New Zealand, and parts of Australia.2 Latitude also correlates with sunlight, as MS risk appears to be inversely related to sunlight exposure and vitamin D.3 Immigration studies also show that migrants who immigrate before puberty acquire the risk profile of their new region, whereas those who immigrate post puberty seem to retain their home region’s risk profile. Although MS is not considered a hereditary disease, genetic factors appear to contribute, with an overall familial recurrence rate of 20%. Monozygotic twin studies show a concordance rate of 24% to 30%, versus 3% to 5% for dizygotic twins. Other risk factors include smoking, vascular comorbidities, and exposure to Epstein-Barr virus in early childhood or mononucleosis after adolescence.3
Clinical Features
MS presents with a variety of neurologic signs and symptoms involving the eyes (acute optic neuritis, diplopia, nystagmus); various muscle groups (facial and limb weakness, spasticity, gait abnormalities, action tremor of the arms, dysarthria, unilateral tonic limb spasms, bladder dysfunction, fecal incontinence, constipation); and the central and peripheral nervous systems (ataxia, paresthesias, fatigue, sexual dysfunction, depression, cognitive impairment, acute pain [e.g., Lhermitte’s sign, ocular pain, dysesthesias], and chronic joint, muscular, and extremity pain).4 In 85% to 90% of cases, disease onset involves a clinically isolated syndrome (CIS) such as optic neuritis, transverse myelitis, or a brainstem or cerebellar syndrome.4 Symptoms may relapse and remit and evolve into progressive disease. Depending on the location of the lesions, symptoms may occur alone or in combination, and as sudden attacks or as part of a steady progression.
As MS progresses, patients experience increased disability over time. Walking impairment is likely to ensue within 10 years, and the need for unilateral support for walking is probable within 15 to 20 years; life expectancy is typically reduced by 7 to 10 years. In 50% of patients, death results from medical complications of the disease; death may also be from suicide or causes similar to those in the general population.1 Indicators of poor prognosis, more rapid disease progression, or rapid conversion from CIS to definite MS are listed in TABLE 1.

Types of MS
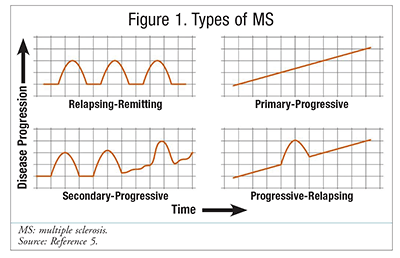
The course of MS can be highly variable from patient to patient; however, there are four recognized types (FIGURE 1): relapsing-remitting MS (RRMS), primary-progressive MS (PPMS), secondary-progressive MS (SPMS), and progressive-relapsing MS (PRMS).5 Initially, most cases are RRMS, which involves periods of neurologic impairment interspersed with periods of recovery that return to baseline with no evidence of disease progression. Over time, neuronal damage becomes clinically evident and the patient typically progresses to SPMS. Most patients with RRMS progress to SPMS after about 10 years, but some may remain in RRMS for 25 years or longer.6 The course of SPMS is characterized by preexisting neurologic deficits that continue to worsen. There are definite periods of relapse, but each relapse typically leads to further disease progression. Approximately 10% of MS patients initially present with PPMS, which is characterized by continuous worsening of disease with few true relapses or plateaus.1 PRMS is somewhat similar to PPMS, but relapses are succeeded by periods of recovery with concomitant progression of MS symptoms.
Diagnosis
The diagnosis of MS is primarily clinical and relies on 1) the signs and symptoms associated with the CNS lesions disseminated in time and space and 2) the exclusion of other disorders that may mimic MS.1 While there is no single diagnostic laboratory test for MS, increased immunoglobulin levels or the presence of two or more oligoclonal bands in the cerebrospinal fluid, delayed latencies of visual, auditory, or somatosensory evoked potential studies, or prolonged central motor conduction times is indicative of demyelination and may represent clinically silent lesions. Blood tests may rule out other diseases. MRI is the most sensitive test for detecting and monitoring white-matter lesions in MS. MRI can be used to support a diagnosis, estimate lesion load and disease activity, assess atrophy and axonal loss, track disease progression, and serve as a surrogate marker of disease. Newer MRI techniques may be used to detect microstructural damage in both white and gray matter.1
Diagnostic criteria for MS have evolved from the original Schumacher criteria (1965) to the McDonald criteria, which appeared in 2001 and were revised in 2005 and 2010.1,7 The original criteria introduced the concepts of dissemination in time (DIT) and dissemination in space (DIS) for lesions, objectively defined relapse in MS, and clarified that the signs and symptoms were not better explained by an alternative diagnosis.1 The first revision of the criteria considered the necessity of early diagnosis and treatment, the utility of MRI in detecting and monitoring MS lesions, and new medications for treating MS. These revisions were made to enhance physicians’ ability to diagnose MS as early and as accurately as possible in patients with a variety of clinical presentations. The 2010 criteria facilitated this goal by simplifying DIS to include at least one lesion in two of four key locations in the CNS and revising DIT criteria to allow for diagnosis from a new lesion on follow-up MRI at any time after baseline, or from a single baseline MRI if it showed two or more demyelinating events, regardless of timing.7 That said, the functional utility of MRI in diagnosing MS is not meant to deemphasize the importance of a thorough clinical examination and differential diagnosis to ensure that there is not a better explanation for the patient’s symptoms.1
Pharmacotherapy
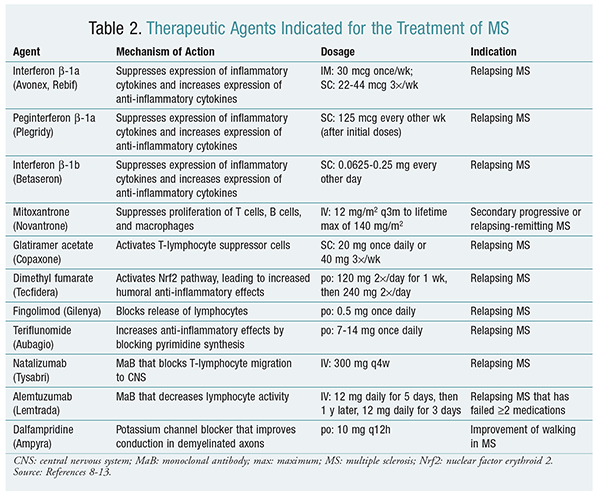
There is currently no cure for MS. The goals of drug treatment are to slow disease progression, limit relapses, reduce long-term neurologic dysfunction, and manage symptoms while limiting adverse reactions. All of the drugs discussed below are approved for RRMS. Other types of MS are typically treated with the same drugs, even though evidence fully supporting their use in these patients is lacking.8,9
Interferon Beta (IFN-β): This agent has been the standard for initial treatment of MS upon diagnosis. Several different preparations of IFN-β are available. IFN-β-1a (Avonex, Rebif) is available in both IM and SC solutions. Peginterferon β-1a (Plegridy) was recently approved by the FDA. IFN-β-1b (Betaseron) is available only in SC form.8 Overall, there is little difference between IFN-β products with regard to efficacy; all tend to reduce relapse rates by about one-third in RRMS patients.9 The most common side effects are injection-site reactions, flulike symptoms, and headache. More serious, but less common, side effects include infections, depression, and increased liver enzymes.8 Unfortunately, IFN-β neutralizing antibodies, flulike symptoms, and the need for self-injection lead to a discontinuation rate of 25% within 1 to 2 years.9 Giving acetaminophen or ibuprofen on administration days may lessen the severity of flulike symptoms.
Glatiramer Acetate (GA; Copaxone): Another commonly used first-line agent is GA, which is administered SC as 20 mg once daily or 40 mg three times a week. Similar to IFN-β preparations, GA reduces the relapse rate by about 35%.8 Injection-site reactions are the most common side effect.
Mitoxantrone (Novantrone): This antineoplastic agent is the only drug indicated for SPMS. It is administered IV at a dosage of 12 mg/m2 every 3 months. Because of the high risk of cardiotoxicity, patients are limited to a lifetime maximum of 140 mg/m2, typically reached after 2 years of treatment. Mitoxantrone is largely reserved for treating SPMS owing to cardiotoxicity, nausea and vomiting, myelosuppression, the risk of secondary leukemia, and the growing plethora of drugs for RRMS.8
Dimethyl Fumarate (DMF; Tecfidera): DMF, a recently approved oral drug for RRMS, is typically begun at 120 mg twice daily for 1 week, then increased to 240 mg twice daily. The lower starting dose is to help alleviate the self-limited effects of flushing and gastrointestinal symptoms. Taking DMF with food or aspirin may reduce the flushing response.9 DMF reduces the relapse rate of RRMS by 40% to 50%.8 DMF has been associated with progressive multifocal leukoencephalopathy (PML), a rare, untreatable, and often fatal disease characterized by a CNS infection caused by the John Cunningham virus (JCV). Anti-JCV serologic testing may be warranted in some patients.9 The most common side effects include flushing, nausea, vomiting, diarrhea, and lymphopenia. Lymphocyte counts should be checked every 6 months.9
Fingolimod (Gilenya): Another oral agent for RRMS is fingolimod. At 0.5 mg once daily, fingolimod has been shown to produce a nearly 50% decrease in RRMS relapse rate and is superior to IFN-β-1a. Fingolimod is contraindicated in patients with previous cardiovascular risk factors (i.e., myocardial infarction, heart failure, arrhythmias, or bradycardia). The first dose of fingolimod should be given in a monitored environment, as bradycardia almost always occurs, although it is rarely symptomatic. Adverse effects include lymphopenia, viral infections, headache, bradycardia, and gastrointestinal symptoms.9
Teriflunomide (Aubagio): Teriflunomide is a metabolite of leflunomide. It is administered orally at either 7 mg or 14 mg daily. The relapse rate is decreased by about 30% with either dosage. The 14-mg dosage has been shown to slow the progression of disability in RRMS patients. Side effects include diarrhea, nausea, vomiting, hair loss, lymphopenia, and elevated liver enzymes.8 Importantly, teriflunomide is a Pregnancy Category X drug. Owing to enterohepatic recirculation, teriflunomide can take up to 2 years to be fully eliminated from the body. In cases of pregnancy, the desire to become pregnant, or an adverse reaction, cholestyramine or activated charcoal may be used over a period of 11 days to hasten the elimination of teriflunomide.9
Natalizumab (Tysabri): This monoclonal antibody (MaB), generally used as second-line therapy for RRMS, is administered IV every 4 weeks.10 Relapse rates have been decreased by up to 68%. Like DMF, natalizumab has been linked to PML, which led to its withdrawal from the market. Natalizumab was reintroduced with a Risk Evaluation and Mitigation Strategy called the TOUCH program.9 A murine component of natalizumab increases the risk of infusion reactions, and the development of anti-natalizumab antibodies decreases the drug’s efficacy.10 Common adverse events include headache, respiratory infections (e.g., pharyngitis), and fatigue.
Alemtuzumab (Lemtrada): Alemtuzumab is a humanized MaB approved for RRMS, and it is administered IV at 12 mg/day on days 1 to 5 of the first year and then for 3 days 1 year later. The annualized relapse rate has been shown to decrease by about 50%.8 Commonly seen adverse effects are headache, diarrhea, and flulike symptoms; there is also a risk of infection and autoimmune thyroiditis.8 Patients should be premedicated with methylprednisolone 1,000 mg for the first 3 days of each treatment period; an antihistamine and an antipyretic may also be considered.
Dalfampridine (Ampyra): Although it is not an MS disease-modifying drug, dalfampridine has been shown to improve walking in MS patients and is approved for this use. Dalfampridine is a potassium channel blocker that is administered at 10 mg twice daily in an extended-release formulation. This agent is associated with seizures, particularly in patients with no prior history of seizure activity. More common adverse reactions include urinary tract infections, headache, dizziness, and insomnia.11
Although there are now many drug choices for lowering the relapse rate of MS, including several oral options, there has been relatively little progress—other than dalfampridine—with regard to treatment of symptoms associated with MS. The mechanisms, indications, and dosages of these drugs are summarized in TABLE 2.8-13
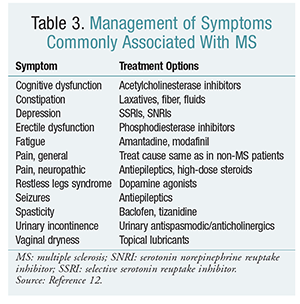
Other symptoms associated with MS are largely treated with the drugs typically used for those symptoms in other situations. TABLE 3 lists symptoms associated with MS and recommended treatment options.
Drugs in Research
A review of ClinicalTrials.gov revealed more than 114 different compounds evaluated in the treatment of MS.14 Three drugs in late-stage clinical development are laquinimod, daclizumab, and ocrelizumab.10 All three are classified as immunologic disease-modifying drugs.
Laquinimod: Laquinimod is a synthetic oral agent that acts by increasing interleukin (IL)-4 and IL-10 production and inhibiting inflammatory-cell infiltration into the CNS. It may also suppress major histocompatibility complex class II antigen-presenting cells and downregulate epitope spreading.10 Laquinimod may also proffer some neuroprotective effects by increasing brain-derived neurotrophic factor. Clinical effects were demonstrated in RRMS in both the ALLEGRO and BRAVO trials, with common side effects including headache, nasopharyngitis, back pain, cough, and depression.10
Daclizumab: This agent is a human MaB that inhibits IL-2–dependent T-cell function and activation of natural killer cells that enter the CNS and suppress pathogenic immune responses. In phase II trials, daclizumab was administered SC every 4 weeks and appeared to be effective as add-on therapy in RRMS. Serious adverse effects were noted, including infections, skin reactions, liver abnormalities, and autoimmune reactions. Phase III development continues.10
Ocrelizumab: Ocrelizumab is a humanized, recombinant MaB that is reactive against B cell–expressed CD20.10 It is administered IV as two doses administered 2 weeks apart every 6 months, resulting in B-cell depletion. In one phase II trial of ocrelizumab versus placebo, ocrelizumab was effective in reducing CNS lesions over 24 weeks, with similar adverse events (other than IV-related events) noted between groups. Phase III trials in RRMS are ongoing.10
Conclusion
The current focus of MS treatment primarily addresses its purported immunologic etiology together with the need for clinical management of associated signs and symptoms. As research leads to a better understanding of MS, it will be important for the pharmacist to stay abreast of the latest developments in the pathophysiology and pharmacotherapy of the disease.
REFERENCES
1. Milo R, Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. 2014;13:518-524.
2. Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev. 2010;9:A387-A394.
3. Kamm CP, Uitdehaag BM, Polman CH. Multiple sclerosis: current knowledge and future outlook. Eur Neurol. 2014;72:132-141.
4. Riley CS, Tullman MJ. Multiple sclerosis. In: Rowland LP, Pedley TA, eds. Merritt’s Neurology. 12th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010:903-918.
5. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology. 1996;46:907-911.
6. Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112:133-146.
7. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292-302.
8. Melzer N, Meuth SG. Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clin Exp Immunol. 2014;175:359-372.
9. Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89:225-240.
10. Du Pasquier RA, Pinschewer DD, Merkler D. Immunological mechanism of action and clinical profile of disease-modifying treatments in multiple sclerosis. CNS Drugs. 2014;28:535-558.
11. Korenke AR, Rivey MP, Allington DR. Sustained-release fampridine for symptomatic treatment of multiple sclerosis. Ann Pharmacother. 2008;42:1458-1465.
12. de Sa JC, Airas L, Bartholome E, et al. Symptomatic therapy in multiple sclerosis: a review for a multimodal approach in clinical practice. Ther Adv Neurol Disord. 2011;4:139-168.
13. Lexi-Drugs [subscription database]. Hudson, OH: Lexi-Comp, Inc; 2015.
14. ClinicalTrials.gov. Multiple sclerosis. www.clinicaltrials.gov/ct2/results?term=multiple+sclerosis&Search=Search. Accessed September 19, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.


