US Pharm. 2013;38(10):HS21-HS24.
ABSTRACT: Pulmonary arterial hypertension (PAH) is a progressively fatal disease of the small pulmonary arteries. In the last two decades, pharmacotherapeutic advances have improved prognosis. Standard treatment of PAH includes anticoagulants, diuretics, digoxin, and calcium channel blockers. Advanced treatment of PAH includes phosphodiesterase type 5 inhibitors, endothelin antagonists, and prostanoids. Combination therapy can produce additive and synergistic benefit. However, drug interactions may occur among PAH therapies or between medications indicated for PAH and agents used for concomitant illnesses. These interactions may necessitate medication adjustment or are serious enough to limit treatment options.
Pulmonary arterial hypertension (PAH) is a complex disorder of the small pulmonary arteries. It is characterized by vascular proliferation and obstructive remodeling, which reduces blood flow through the pulmonary arterial circulation, increases pulmonary vascular resistance, and eventually causes right heart failure. The hemodynamic definition of PAH is a resting mean pulmonary arterial pressure (mPAP) of ≥25 mmHg and a pulmonary capillary wedge pressure (PCWP) of ≤15 mmHg.1,2
Epidemiology
PAH is a rare and fatal disease estimated to affect 10 to 12 per one million Americans.3 Risk factors include connective tissue diseases, congenital heart disease, portal hypertension, HIV infection, and certain drugs and toxins (e.g., aminorex, amphetamines, fenfluramine, dexfenfluramine, L-tryptophan, toxic rapeseed oil).4 Obesity and use of cocaine and amphetamines may be emergent risk factors.5 Patients with PAH have a mean age of 50 years and are predominantly female.5 Traditionally, the mean survival time is 2.8 years, but prognosis has improved with recent effective therapies.
Classification
The current World Health Organization (WHO) Group Dana Point classi-fication delineates PAH as follows: idiopathic; heritable; associated with collagen vascular disease; congenital systemic-to-pulmonary shunts; portal hypertension; HIV; schistosomiasis; drugs and toxins; hereditary hemorrhagic telangiectasia; hemoglobinopathies; associated with significant venous or capillary involvement, pulmonary veno-occlusive disease, and pulmonary capillary hemangiomatosis; and persistent pulmonary hypertension of the newborn.4,6 Chronic hemolysis was removed from Group 1 during the 2013 Pulmonary Hypertension World Symposium in Nice, France.
Pathophysiology
Genetic mutations (BMPR2, ACVRL1, ALK1), signaling pathway defects (TGF-beta) and dysregulation of receptor and transporter activities (HTR2B, SLC6A4, KCNA5, KCNQ, TRPC6, ANGPT ), have been implicated in the development of PAH.7-12 These abnormalities can produce an imbalance of smooth muscle proliferation and cellular apoptosis that favors hyper-trophy and plexiform lesions. In addition, in situ thromboses further obliterate the vascular lumen.
Clinical Presentation and Diagnosis
The main symptoms of PAH are dyspnea and exercise limitation. Other symptoms include fatigue, angina, syncope, palpitations, lightheadedness, and abdominal distention. Patients with early onset PAH may be asymptomatic, and diagnosis is made by exclusion of other diseases, such as chronic obstructive pulmonary disease. PAH is strongly suspected when symptoms occur in the absence of cardiac or respiratory disease or when they cannot be explained by preexisting disease. Patients with advanced PAH may additionally present with cold extremities, hypotension, cardiac murmurs, and reduced pulse pressure. Ultimately, right heart catheterization is needed to make a definitive diagnosis.4
Stages of PAH
The New York Heart Association (NYHA) classification has been adapted by the WHO to define the clinical stages of PAH. Class I patients have no limitations to physical activity. Patients with class II functional activity are comfortable at rest; however, routine activities can cause undue dyspnea, fatigue, chest pain, or syncope. With class III status, patients have a marked decline in ability to carry out daily routines. Although still comfortable at rest, class III patients experience symptoms when performing less-than-ordinary activities. Finally, patients with class IV functional status have significant dyspnea, fatigue, chest pain, or syncope to the extent that they are unable to carry out routine activities.13 These patients also exhibit other symptoms of heart failure, such as lower extremity edema, palpitations, arrhythmia, tachycardia, and nocturia.
PAH Treatment
The goals of PAH therapy are to improve symptoms, enhance quality of life, and slow disease progression.14 Standard treatment with diuretics, digoxin, anticoagulants, and oxygen is recommended for most patients with PAH. Calcium channel blockers (CCBs) are recommended for patients with PAH when they respond positively to acute vasodilator testing, which is defined as a decrease in mPAP of ≥10 mmHg to an absolute value of <40 mmHg without decreased cardiac output.14 Therapies for advanced care include phosphodiesterase type 5 (PDE5) inhibitors, endothelin receptor antagonists, and prostanoids.
Combining agents may produce additive therapeutic benefit or allow for similar benefit with lower doses and fewer side effects. However, combination therapy increases the potential for drug interactions among medications for PAH or between medications for PAH and those for concurrent diseases such as hypertension, cardiovascular disease, depression, obstructive airway disease, and thyroid disease.5 Drug interactions may warrant medication adjustments or limit treatment choices. Here, we summarize the drug interaction potential of PAH therapy.
Drug Interactions of Standard Treatments for PAH
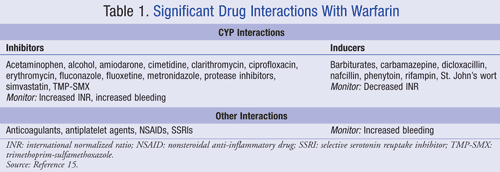
Warfarin: Patients with PAH have increased risk for thromboembolism. Warfarin (Coumadin, Jantoven) is used to prevent and treat arterial thromboembolism, and in retrospective studies has been shown to confer survival benefit.14 It is recommended that warfarin therapy be titrated to an international normalized ratio (INR) of 1.5 to 2.5.14 Given its significant interactions with other CYP-mediated medications, close monitoring of INR is warranted.15,16 Selected warfarin interactions are presented in TABLE 1.15
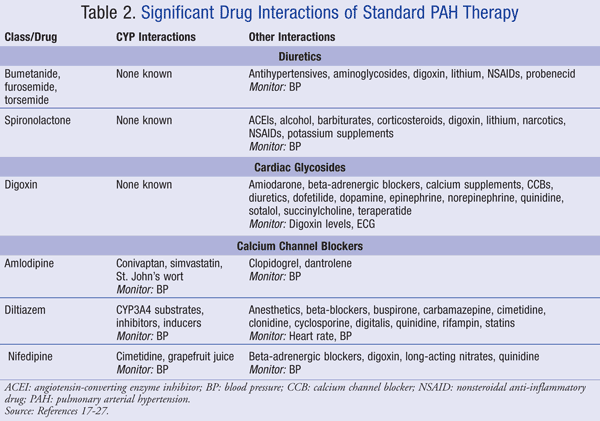
Diuretics: Diuretics are used to manage edema and right ventricle volume overload. The major interactions of loop diuretics exist with agents that can cause nephrotoxicity, ototoxicity, electrolyte disturbances, or additive hypotension.17-22 Spironolactone (Aldactone) is a potassium-sparing diuretic that is frequently used for PAH. It has been shown to effectively block aldosterone and decrease atrial natriuretic peptide.23
Digoxin: Digoxin can improve cardiac output and help with atrial arrhythmias. Digoxin is a substrate for P-glycoprotein (Pgp); thus, concomitant use with drugs that affect Pgp may alter digoxin levels. Serum digoxin levels should be established before starting new medications and monitored thereafter with appropriate dosage adjustment.24
Calcium Channel Blockers: CCBs may have a potential vasodilatory effect. They are usually used in patients with idiopathic PAH who exhibit a positive response to acute vasodilator testing. This is defined as a ≥10 mmHg decrease in mPAP to an absolute mPAP <40 mmHg without reduction in cardiac output.14 Nifedipine (Procardia, Adalat), diltiazem (Cardizem, Dilacor), and amlodipine (Norvasc) are the most commonly used CCBs for PAH.25-27
A summary of drug interactions of standard treatments for PAH is presented in TABLE 2.17-27
Drug Interactions of Advanced Treatments for PAH
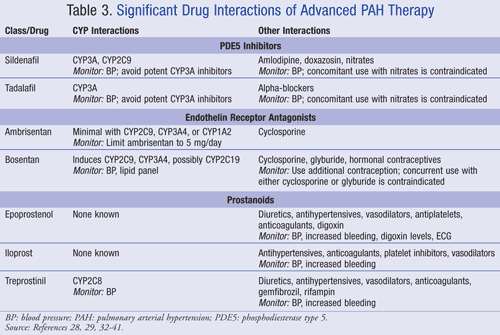
PDE5 Inhibitors: PDE5 inhibitors cause nitric oxide-mediated vasodilation. Together, PDE5 inhibitors and nitrates can produce profound hypotension, and this combination is contraindicated. Additive hypotension can also occur with alpha-blockers.28,29 Concurrent use of sildenafil with potent CYP3A inhibitors is not generally recommended.30 Use of sildenafil (Viagra) and tadalafil (Cialis) should be avoided with other PDE5 inhibitors.
Endothelin Receptor Antagonists: Endothelin-1 (ET-1) is a potent vasoconstrictor and stimulator of pulmonary arterial smooth muscle cell proliferation. Plasma levels of ET-1 are correlated with increased severity and worsened prognosis of PAH. Endothelin receptor antagonists, such as bosentan (Tracleer) and ambrisentan (Letairis), work by competitively inhibiting ET-1 from binding endothelin receptors to decrease pulmonary vascular resistance.14 Bosentan has been shown to have more drug interactions than ambrisentan.31 Cyclosporine is contraindicated with bosentan, and efficacy of hormonal contraception may be reduced with bosentan.32,33 Concomitant use of bosentan and glyburide is also contraindicated due to their CYP3A4-inducing potential, which results in increased plasma levels of both drugs.32,34 When bosentan and sildenafil are given concurrently, bosentan levels may increase and sildenafil levels may decrease, although dosage adjustments may not be necessary.32 To date, ambrisentan is associated with one clinically significant interaction. If used with cyclosporine, ambrisentan should be limited to 5 mg daily.35,36
Given the risk for liver injury and teratogenic harm (Pregnancy Category X), bosentan and ambrisentan are only available through the Tracleer Access Program and Letairis Education and Access Program, respectively.32,36
Prostanoids: Prostacyclin synthase is reduced in patients with PAH. As a result, there is insufficient production of prostacyclin (PGI2), a vasodilator with antiproliferative effects.14 Patients with PAH are treated with prostanoids to replace the prostacyclin within the pulmonary arterial vasculature.37 Prostanoids induce pulmonary arterial vasodilation, impede smooth muscle cell growth, and disrupt platelet aggregation.37
Several prostanoids are available. Iloprost (Ventavis) is inhaled using the I-neb AAD System or the Prodose AAD System.37,38 Treprostinil (Tyvaso) is another prostanoid that can be administered by inhalation. To date, there are no pharmacokinetic studies with inhaled treprostinil. Drug interaction potential is based upon what is known with oral or injected treprostinil (Remodulin). Prostanoids available for parenteral administration include epoprostenol (generic, Flolan, Veletri) and trepostinil (Remodulin). Epoprostenol is delivered as a continuous IV infusion pump due to its short half-life of 2.7 minutes.39 It was the first widely used prostanoid and has been shown to effectively dilate pulmonary arteries, reduce symptoms of PAH, and provide survival benefit.37 Treprostinil can be given as an SC infusion, or an as IV infusion for patients who experience intolerable irritation from SC administration. Dosage adjustment of trepostinil may be necessary when it is used with gemfibrozil or rifampin.40,41
A summary of drug interactions of advanced treatments for PAH is presented in TABLE 3.28,29,32-41
Role of the Pharmacist
Treatment for PAH involves a complicated medication regimen. Pharmacists in hospital and community settings can help patients understand the need for different types of medications and nonpharmacologic therapies. By appropriately applying information from package labeling, medication databases, postmarketing reports, and FDA MedWatch notices, pharmacists can help to determine the clinical relevance of drug interactions of PAH therapies and the need for medication adjustments. Pharmacists can also integrate other medications into the pharmacotherapy plan.
Although PAH is a rare and fatal disease, prognosis has improved with expanding therapeutic options. New medications are in development, such as Selexipag (ACT-293987).42 Pharmacists with specialized knowledge about medications are well positioned to perform drug monitoring, which can prevent adverse events and ultimately optimize patient care.
REFERENCES
1. Badesch DB, Abman SH, Simonneau G, et al. Medical
therapy for pulmonary arterial hypertension: updated ACCP evidence-based
clinical practice guidelines. Chest. 2007;131:1917-1928.
2. Hoeper MM. Definition, classification, and epidemiology of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:369-375.
3. Frost AE, Badesch DB, Barst RJ, et al. The changing
picture of patients with pulmonary arterial hypertension in the United
States: how REVEAL differs from historic and non-US Contemporary
Registries. Chest. 2011;139:128-137.
4. Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2009;30:2493-2537.
5. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary
arterial hypertension: baseline characteristics from the REVEAL
registry. Chest. 2010;137:376-387.
6. Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S43-S54.
7. Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation. 2005;111:534-538.
8. Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous
germline mutations in BMPR2, encoding a TGF-beta receptor, cause
familial primary pulmonary hypertension. Nat Genet 2000;26:81-84.
9. Deng Z, Morse JH, Slager SL, et al. Familial primary
pulmonary hypertension (Gene PPH1) is caused by mutations in the bone
morphogenetic protein receptor–II gene. Am J Hum Genet. 2000;67:737-744.
10. Girerd B, Montani D, Coulet F, et al. Clinical
outcomes of pulmonary arterial hypertension in patients carrying an
ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181:851-861.
11. Harrison RE, Berger R, Haworth SG, et al. Transforming
growth factor-beta receptor mutations and pulmonary arterial
hypertension in childhood. Circulation. 2005;111:435-441.
12. Gurney AM, Joshi S, Manoury B. KCNQ potassium channels: new targets for pulmonary vasodilator drugs? Adv Exp Med Biol. 2010;661:405-417.
13. Kuhr FK, Smith KA, Song MY, et al. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2012;302:H1546-H1562.
14. McLaughlin VV, Archer SL, Badesch DB, et al. ACCP/AHA 2009 expert consensus document on pulmonary hypertension. J Am Coll Cardiol. 2009;53:1573-1619.
15. Coumadin (warfarin) package insert. Princeton, NJ: Bristol-Myers Squibb Co; October 2011.
16. Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111-117.
17. Bumex (bumetanide) package insert. Parsippany, NJ. Validus Pharmaceuticals LLC; September 2009.
18. Lasix (furosemide) package insert. Bridgewater, NJ: Sanofi-Aventis LLC; August 2011.
19. Demadex (torsemide) package insert. Somerset, NY: Meda Pharmaceuticals, Inc; February 2010.
20. Aldactone (spironolactone) package insert. New York, NY: Pfizer, Inc; January 2008.
21. Micromedex [online subscription database]. Greenwood
Village, CO: Truven Health Analytics, Inc. Updated periodically.
www.micromedexsolutions.com. Accessed March 16, 2013.
22. Lexicomp [online subscription database]. Hudson, OH:
Lexi-Comp, Inc. Updated periodically. http://online.lexi.com. Accessed
March 16, 2013.
23. Pitt B, Zannad F, Remme WJ. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999:341:709-717.
24. Digoxin package insert. Columbus, OH: Roxane Laboratories, Inc; September 2012.
25. Procardia XL (nifedipine) package insert. New York, NY: Pfizer, Inc; August 2011.
26. Cardizem (diltiazem) package insert. Bridgewater, NJ: BTA Pharmaceuticals, Inc; November 2009.
27. Norvasc (amlodipine) package insert. Research Triangle Park, NC: Synthon Pharmaceuticals, Inc; September 2007.
28. Revatio (sildenafil) package insert. New York, NY: Pfizer Inc; August 2012.
29. Adcirca (tadalafil) package insert. Indianapolis, IN: Eli Lilly and Company; March 2012.
30. Muirhead GJ, Wulff MB, Fielding A, et al. Pharmacokinetic interactions between sildenafil and saquinavir/ritonavir. Br J Clin Pharmacol. 2000;50:99-107.
31. Weiss J, Herzog M, Haefeli WE. Differential modulation
of the expression of important drug metabolizing enzymes and
transporters by endothelin-1 receptor antagonists ambrisentan and
bosentan in vitro. Eur J of Pharmacol. 2011;660:298-304.
32. Tracleer (bosentan) package insert. South San Francisco, CA: Actelion Pharmaceuticals; October 2012.
33. Treiber A, Schneiter R, Hausler S, et al. Bosentan is a
substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as
the common mechanism of its interactions with cyclosporine A,
rifampicin, and sildenafil. Drug Metab Dispos. 2007;35:1400-1407.
34. van Giersbergen PL, Treiber A, Clozel M, et al. In
vivo and in vitro studies exploring the pharmacokinetic interaction
between bosentan, a dual endothelin receptor antagonist, and glyburide. Clin Pharmacol Ther. 2002;71:253-262.
35. Frampton JE. Ambrisentan. Am J Cardiovasc Drugs. 2011;11:215-226.
36. Letairis (ambrisentan) package insert. Foster City, CA: Gilead Sciences; October 2012.
37. Bishop BM, Mauro VF, Khouri SJ. Practical considerations for the pharmacotherapy of pulmonary arterial hypertension. Pharmacotherapy. 2012;32:838-855.
38. Ventavis (iloprost inhalation solution) package insert. South San Francisco, CA: Actelion Pharmaceuticals; August 2012.
39. Veletri (epoprostenol for injection) package insert. South San Francisco, CA: Actelion Pharmaceuticals; June 2012.
40. Tyvaso (trepostinil inhalation solution) package
insert. Research Triangle Park, NC: United Therapeutics Corp; February
2011.
41. Remodulin (trepostinil injection) package insert. Research Triangle Park, NC: United Therapeutics Corp; February 2011.
42. Selexipag (ACT-293987) in pulmonary arterial
hypertension, GRIPHON trial. ClinicalTrials.gov.
http://clinicaltrials.gov/show/NCT01106014. Accessed September 16, 2013.
To comment on this article, contact rdavidson@uspharmacist.com.


