US Pharm. 2007;32(4):HS29-HS39.
M.T. is a 30-year-old woman who presents to her lawyer at six-weeks gestation. She states she was diagnosed 10 weeks ago with lupus vulgaris on the face, a progressive form of cutaneous tuberculosis. For treatment, her physician prescribed rifampin in combination with isoniazid. She states the only medication she was taking at that time was a low-dose birth control pill (one ethinyl estradiol/levonorgestrel); however, one month after starting rifampin and isoniazid, she discovered that she was pregnant. She is now considering a lawsuit. She claims that her pharmacist and physician failed to inform her that there is a known drug interaction between rifampin and low-dose ethinyl estradiol that would increase her risk of ovulation and result in pregnancy. Neither the pharmacist nor the physician had recommended an alternative contraceptive or a higher dose of estrogen.1
Pharmacists face many challenges regarding drug interactions, since some interactions do not produce harmful effects, while others lead to life-threatening events or death. Drug interactions have become an important safety concern, causing up to 2.8% of all hospital admissions.2 Due to a significant rise in polypharmacy, clinicians have become increasingly concerned about drug interactions.2 In addition, a 2002 survey showed that patients rank drug interactions as their greatest concern over any other health issue in a hospital or health system.3 This article aims to assist pharmacists in identifying patients at risk for clinically significant dermatologic drug interactions, review the general mechanisms of drug interactions, and help lower patients' risk with a discussion of select dermatologic drugs that may lead to adverse effects. With this knowledge, as well as proper screening, prescribing, and monitoring, clinicians can help prevent fatal consequences.
A drug interaction occurs when one drug (the precipitant drug) alters the pharmacologic effect of another drug (the object drug) when both are given concurrently. The drug interaction may cause either enhanced or diminished activity of the object drug, resulting in toxicity or therapeutic failure, respectively.4 During drug development, investigators report incomplete information about drug interactions when investigating select cytochrome P-450 (CYP) isoenzymes in healthy patients using two-drug combinations.2 Clinical interpretation is difficult because pharmacists cannot extrapolate the data to chronic or acutely ill patients who receive multiple medications with opposing effects on metabolism.
Although it is impossible for clinicians to remember all potential drug interactions, knowledge of the pharmacokinetic properties of "red flag drugs" or drug classes can lower the risk of adverse drug reactions.
Patients at Risk
Drug interactions are most prevalent in the elderly, neonates,
immunocompromised patients, patients with psychiatric conditions, and those
with renal and liver disorders. Polypharmacy (taking excessive medications
with inappropriate use) and patients who have multiple prescribers or "doctor
shoppers" with poor prescriber communications can raise the risk.4,5
Genetics also has a role, as polymorphic genes cause certain enzymes, such as
CYP2D6, to be less effective. Gender differences affect drug absorption,
gastric emptying, and drug distribution, whereas obesity reduces CYP3A4
metabolism.2,6 Drug interactions may be of minor significance in a
healthy individual, but the variety of these influential factors can severely
exacerbate chronic conditions.4
Mechanism of Action
Drug interactions can be described as pharmacodynamic or pharmacokinetic.
Pharmacodynamic drug interactions are additive, synergistic, or antagonistic
combinations, where the pharmacologic effect of the drug is changed.4
For example, such drug combinations as aspirin and warfarin can increase
anticoagulation as an additive effect. In dermatology, most of the drug
interactions are pharmacokinetic reactions. Pharmacokinetic drug interactions
occur when the precipitant drug affects the absorption, distribution,
metabolism, or excretion of the object drug.4
Pharmacokinetic drug interactions occur in the liver where lipophilic drugs are metabolized to hydrophilic drugs and are excreted by the kidneys. Drug metabolism consists of phase I and phase II reactions. Phase I, involving hydroxylation, oxidation, and reduction, is catalyzed by the CYP-450 family, whereas phase II involves glucuronidation and conjugation, which produce a more polar and less toxic compound.4
Absorption: The risk of an interaction within the gastrointestinal tract is influenced by gastric pH, intestinal motility, and agents that form insoluble compounds with the object drug.2 Ketoconazole and itraconazole, best absorbed in an acidic environment, can be affected by drugs that increase gastric pH, such as antacids, histamine (H2) receptor blockers, and proton pump inhibitors. In addition, multivalent cations such as magnesium and aluminum in antacids bind to tetracycline and fluoroquinolones and form an insoluble, inefficacious compound.2
Distribution: Highly protein-bound drugs can cause drug interactions when the precipitant drug displaces the object drug from the plasma protein-binding sites, resulting in increased serum concentration of the free (object) drug. However, since the free drug is eliminated more readily, this may not result in a clinically important drug interaction.2 A protein-binding displacement is clinically significant if the object drug has a narrow therapeutic index, has limited distribution in the body, and is slowly eliminated. An important protein-binding drug interaction is methotrexate (MTX) and nonsteroidal anti-inflammatory drugs (NSAIDs); NSAIDs not only displace MTX from binding sites but also reduce its renal elimination.2 Increased MTX serum levels may result in MTX toxicity (e.g., bone marrow depression, gastrointestinal effects).
Metabolism: One important mechanism of drug interactions is metabolism, which consists of drug clearance changes involving the CYP-450 isoenzyme system. Hepatocytes and enterocytes contain the highest concentrations of CYP enzymes. Over 90% of drug oxidation can be attributed to six main cytochromes: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. Drugs act as substrates, inhibitors, or inducers of specific isoenzymes.2 A substrate is metabolized by an enzyme at the enzymatic-binding site. An enzyme inhibitor competes with the substrate for the enzyme-binding sites, decreasing the substrate's ability to be metabolized and resulting in an increased serum level of the substrate. For example, when warfarin is given concomitantly with fluconazole, a potent inhibitor of CYP2C9, fluconazole will inhibit warfarin's metabolism, causing increased prothrombin time and potential risk of bleeding. In contrast, an enzyme inducer stimulates the production of increased liver enzymes, increasing the metabolism of the substrate and resulting in decreased levels of the substrate. For example, when warfarin is given concomitantly with rifampin, a potent CYP2C9 inducer, rifampin will induce warfarin's metabolism, causing decreased warfarin levels and leading to subtherapeutic prothrombin times with the risk of a blood clot.
When a drug reaches the intestinal enterocytes, a variety of plasma membrane transporters such as P-glycoprotein (P-gp) are responsible for metabolism. P-gp expels drug back into the intestinal lumen to limit absorption of toxic lipophilic substances. Ketoconazole and itraconazole are both substrates and inhibitors of P-gp, making the effect of this interaction unpredictable.7
Excretion: A drug can affect the renal clearance of another drug by altering its excretion, leading to increased serum concentrations. For example, when penicillin and probenecid are concomitantly administered, probenecid competitively inhibits the renal secretion of penicillin, increasing penicillin's concentration and prolonging its activity.
Drug Interactions of Clinical
Significance
This article focuses on drug interactions involving specific drugs and drug
classes of clinical significance in dermatology, including MTX, cyclosporine
A, terbinafine, pimozide, azole antifungals, macrolide antibiotics, quinolone
antibiotics, and rifampin (see also Table 1).
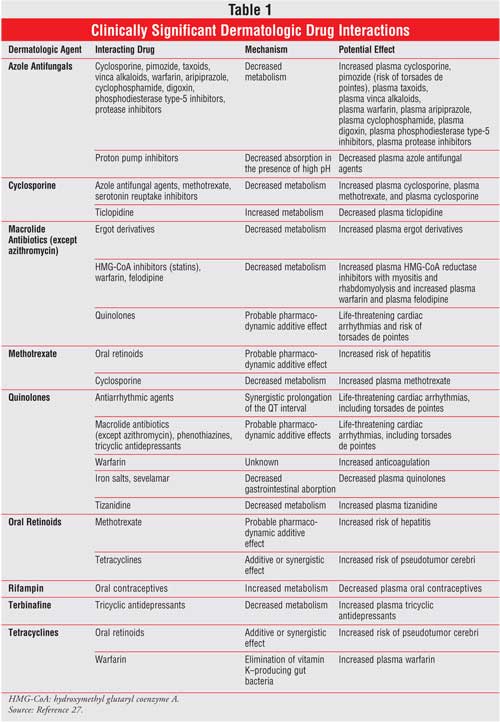
Methotrexate: MTX is used for the treatment of recalcitrant psoriasis.8 It is 35% metabolized by the enterohepatic system after oral administration and primarily renally cleared by glomerular filtration and active tubular secretion.5,9 Patients with impaired renal function can experience markedly increased MTX serum levels.
When MTX and penicillins are coadministered, penicillins compete with the renal tubular secretion of MTX by inhibiting the cellular uptake of MTX and delaying its clearance, causing MTX-induced neutropenia, renal failure, and mucositis.5,10,11 However, the risk with low-dose MTX (7.5 to 25 mg/day) is probably much lower than the risk when using MTX at antineoplastic dosages (500 mg to 1.2 g/m2 /week).1 An alternative antibiotic may be warranted. In addition, clinicians should avoid concomitant use of probenecid, which can increase MTX levels by two- to threefold.1
The renal clearance of MTX is significantly reduced with concurrent administration of choline magnesium trisalicylate, ibuprofen, or naproxen. Both trisalicylate and ibuprofen reduce MTX renal clearance significantly, but trisalicylate displaces MTX from protein, increasing the free MTX by 28%.12 Pharmacists may consider recommending acetaminophen, ketoprofen, flurbiprofen, or piroxicam, since these agents do not affect MTX disposition.1,13
Some investigators suggest avoiding the combination of MTX and cyclosporine for psoriasis due to toxic side effects, while other studies show minimal adverse effects with low dosages and short-term therapy (<1 year).8,14 Clinicians must monitor patients for MTX toxicity, evaluate the patient's renal clearance and MTX efficacy with interacting drugs, and consider alternative therapy if necessary.
Cyclosporine A: Cyclosporine A is used for the treatment of severe psoriasis and is metabolized by P-gp in the intestine and CYP3A4 isoenzymes in the liver. 2,4,5,8 Due to its narrow therapeutic index, CYP inducers and inhibitors affect serum concentrations, resulting in loss of efficacy or toxicity, respectively.2,4,5 Enzyme inhibitors include macrolide antibiotics (erythromycin, clarithromycin, telithromycin), azole antifungals (fluconazole, itraconazole, ketoconazole), calcium channel blockers (nicardipine, diltiazem, felodi pine, verapamil), antivirals (amprenavir, atazanavir, ritonavir, saquinavir, delavirdine), and grapefruit juice.1 Clinicians intentionally use CYP3A4 inhibitors to increase cyclosporine's bioavailability, which allows for reduced cyclosporine dosages, decreased adverse effects, and lower cost.2 Coadministration with ketoconazole 200 to 400 mg daily can decrease cyclosporine dosage by 60% to 80%, while diltiazem can decrease the dosage by 30%. Grapefruit juice can also decrease the dosage, but the effects are variable and not recommended.2 Cyclosporine inducers include St. John's wort, rifabutin, primidone, nevirapine, efavirenz, phenobarbital, phenytoin, rifampin, and carbamazepine. 1,2 Clinicians should monitor cyclosporine trough levels to ensure adequate therapeutic response and use alternate therapy if needed. Patients should be counseled to avoid grapefruit juice, which inhibits cyclosporine metabolism.
Terbinafine: Terbinafine is used to treat dermatophytoses.2 Oral terbinafine is a potent inhibitor of CYP2D6 isoenzymes and inhibits the metabolism of beta-blockers, such as carvedilol, metoprolol, timolol, and propranolol, causing significant beta blockade that may lead to hypotension, bradycardia, or heart failure.1,2,15 Clinicians may use beta-blockers that are not metabolized by CYP2D6 (e.g., atenolol or nadolol), avoid terbinafine use, and use an alternative antifungal or alternative drug class.1
Terbinafine inhibits the metabolism of codeine to its active metabolite (morphine), causing codeine to lose its analgesic effect.2 To prevent this interaction, analgesics can be used that do not require metabolism by CYP2D6, such as morphine, methadone, fentanyl, hydromorphone, and oxycodone.1 Pharmacists can recommend terbinafine as an alternative to azole antifungals for the treatment of onychomycosis if the patient is concurrently taking a drug that is a substrate of CYP3A4 isoenzymes.
Pimozide: Pimozide is a potent, long-acting neuroleptic used to treat psychogenic dermatologic problems, such as delusions of parasitosis, where the patient believes the diagnosis is dermatologic rather than psychiatric.16,17 Pimozide, metabolized to a greater extent by CYP3A4 and to a lesser extent by CYP1A2, is a powerful inhibitor of CYP2D6, although it is not a substrate of this isoform. 16 Pimozide has been shown to cause prolongation of the QT interval associated with fatal ventricular arrhythmias (torsades de pointes), grand mal seizures, and unexpected death.16-18 Flockhart et al. reported two cases of fatal ventricular arrhythmias from the concomitant administration of pimozide with clarithromycin.18 They studied this combination further in healthy individuals who were poor or extensive metabolizers of the CYP2D6 system.19 Results indicated that clarithromycin inhibits pimozide metabolism, and pimozide prolongs the QT interval, causing cardiac toxicity.
Pimozide is contraindicated in patients using protease inhibitors (e.g., indinavir, nelfinavir, saquinavir, ritonavir), CYP3A4 inhibitors (e.g., clarithromycin, azole antifungals), CYP1A2 inhibitors (e.g., macrolides, fluvoxamine, cimetidine, fluoroquinolones), sertraline, and grapefruit juice.1,16 CYP3A4 inhibitors should not be used concomitantly with pimozide due to the risk of life-threatening ventricular arrhythmias; alternative therapy is recommended. Pimozide is contraindicated in patients who are at risk of ventricular arrhythmias, prolonged QT intervals, or electrolyte disturbances.16 Electrolyte disturbances can occur with concomitant use of drugs that affect QT intervals, such as phenothiazines, tricyclic antidepressants, antiarrhythmics (dofetilide, sotalol, quinidine, class IA and class III antiarrhythmics), thioridazine, droperidol, moxifloxacin, pentamidine, and tacrolimus.16
Azole Antifungals: In dermatology, azole antifungals are used most commonly to treat superficial mycoses, including tinea versicolor, tinea capitis, tinea corporis, tinea cruris, tinea pedis, and onychomycoses. Azoles are substrates and inhibitors of P-gp and CYP3A4 isoenzymes.7 Ketoconazole has been shown to be the strongest inhibitor of the CYP3A4 system. Itraconazole is also an inhibitor of CYP3A4, whereas fluconazole is a greater inhibitor of CYP2C9 than CYP3A4. Drugs of clinical importance that interact with azoles include warfarin, phenytoin, and cyclosporine.20 Fluconazole inhibits the metabolism of warfarin, leading to increased prothrombin time and risk of bleeding. Phenytoin is metabolized by CYP2C9, and fluconazole has been reported to decrease its clearance by 33%.2 Two cases of phenytoin toxicity have been reported when phenytoin was concomitantly administered with bedtime fluconazole at 200 mg/day.21 Therefore, clinicians must frequently monitor prothrombin times, phenytoin levels, or cyclosporine levels for patients taking azoles in combination with warfarin, phenytoin, or cyclosporine therapy, respectively.
Azoles interfere with the metabolism of benzodiazepines, such as triazolam and midazolam, leading to increased benzodiazepine serum levels; concomitant use of azoles with statins has led to rhabdomyolysis. Itraconazole is contraindicated for use with triazolam, midazolam, lovastatin, simvastatin, astemizole, quinidine, and pimozide.22 Itraconazole can be used with an alternative statin (e.g., fluvastatin).
In addition, the azoles require an acidic environment for absorption. Thus, concomitant use with antacids, H2 -receptor antagonists, proton pump inhibitors, sucralfate, and didanosine reduces absorption significantly.2,20
According to one study, 70% of patients admitted to a hospital who were taking azole antifungals experienced azole drug interactions.23 Most of these interactions occurred with moderate to major severity with the coadministration of prednisone, midazolam, warfarin, methylprednisolone, cyclosporine, and nifedipine.
Macrolide Antibiotics: Macrolides have been used for the treatment of acne vulgaris, rosacea, erythrasma, pity riasis lichenoid, impetigo, and boils. The macrolides, especially erythromycin, are potent inhibitors of CYP3A4 isoenzymes.24 However, unlike erythromycin and cla rithromycin, dirithromycin and azithromycin are not inhibitors of the CYP3A4 isoenzymes. Substrates of the CYP3A4 isoenzymes, such as warfarin, tacrolimus, silden afil, and carbamazepine, may significantly interact with erythromycin and clarithromycin. 1
In most cases, the interaction of erythromycin and carbamazepine has led to carbamazepine toxicity (nausea, vomiting, dizziness, diplopia, headache, and confusion) within two to three days of coadministration. If concurrent administration of these drugs cannot be avoided, a lower carbamazepine dosage may be prudent.1
Quinolone Antibiotics: These antibiotics have been used to treat skin and skin structure infections caused by Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Proteus mirabilis, Proteus vulgaris, Providencia stuartii, Morganella morganii, Citrobacter freundii , Pseudomonas aeruginosa, Staphylococcus species (methicillin susceptible), Staphylococcus epidermidis, and Streptococcus pyogenes .25
Multivalent cations (e.g., antacids containing magnesium, aluminum, and, to a lesser extent, calcium compounds) interact with fluoroquinolones and reduce their absorption, with subsequent decreased bioavailability.1,25 Ciprofloxacin, norfloxacin, and moxifloxacin are more affected than levofloxacin.1,2 Patients should take the quinolone two hours before or six hours after the cations to minimize this interaction.25 Bioavailability also decreases with sucralfate; this combination should be avoided, or the quinolone should be taken two to six hours prior to sucralfate administration.1
Rifampin: Rifampin treats cutaneous tuberculosis, leprosy, pyoderma, leishmaniasis, psoriasis, and pruritus.26 Rifampin is a potent inducer of CYP2C9, CYP2C19, and CYP3A4 enzymes and decreases the serum concentrations of such drugs as warfarin, cyclosporine, theophylline, digoxin, methadone, ketoconazole, phenytoin, and oral contraceptives.4,26 Rifampin exerts its effects three weeks after the initiation of therapy, and the drug interaction may persist for up to four weeks after rifampin discontinuation. Alternative therapy should be given, and clinicians should avoid the concomitant administration of these substrates. Clinicians should counsel patients to choose an oral contraceptive with higher estrogen content (>35 mcg of ethinyl estradiol) or an alternative contraceptive in combination with rifampin.1,4,26
Role of the Pharmacist
M.T. (from the patient case introduced earlier) has experienced a
well-documented drug interaction with concomitant use of rifampin and oral
contraceptives. The physician and pharmacist were responsible to counsel the
patient and advise her to use an alternative form of contraception.
To decrease the risks of drug interactions, pharmacists should continuously update the patient's medication profile, such as use of herbals, OTCs, and natural supplements, and monitor for red-flag drugs or drugs with a narrow therapeutic index. Pharmacists must evaluate the magnitude of change expected in drug plasma concentrations based on the patient's risk factors, multiple medications, interacting drugs, and clinically significant reactions. It is advisable to avoid the interaction if the drugs are contraindicated, circumvent the interaction where necessary, and use an alternative therapy if possible. The pharmacist should also ensure the drug is being used for a well-documented, beneficial drug interaction. (For example, the combination of cyclosporine and diltiazem can significantly reduce cost and renal toxicity, but the patient must have periodic serum cyclosporine levels monitored to ensure therapeutic cyclosporine levels in order to prevent organ rejection and renal toxicity.)
The pharmacist has a responsibility to warn patients and prescribers about drug interactions. The pharmacist should have knowledge about interactions and not depend solely on drug-interaction databases that screen or monitor a patient's medication profile, since these databases are not always reliable. Clinicians should report observed drug interactions to regulatory bodies such as the FDA (via Medwatch) and submit case reports in journals. Patients can reduce their risk by using only one pharmacy to obtain their drugs and informing every clinician of all their current medications.
Pharmacists should keep abreast of current literature and utilize valuable resources, such as The Medical Letter's Handbook on Adverse Drug Interactions and Hansten and Horn's Top 100 Drug Interactions. Hopefully, with medication therapy management services through Medicare Part D, pharmacists will get involved and be vigilant in screening medications for clinically significant drug interactions.
References
1. Hansten PD, Horn JR. The Top 100 Drug Interactions: A Guide to Patient
Management. 2006 Edition. Freeland, WA: H&H Publications; 2006.
2. Shapiro LE, et al. Drug interactions of clinical significance for the
dermatologist. Recognition and avoidance. Am J Clin Dermatol.
2003;4:623-639.
3. ASHP. Survey reveals patient concerns about medication related issues.
Available at:
www.ashp.org/s_ashp/sec_press_article.asp?CID=168&DID=2037&id=2996. Accessed
January 13, 2007.
4. Aria N, Kauffman L. Important drug interactions and reactions in
dermatology. Dermatol Clin. 2003;21:207-215.
5. Andersen WK, Feingold DS. Adverse drug interactions clinically important
for the dermatologist. Arch Dermatol. 1995;131:468-473.
6. Kolyar M, Carson SW. Effects of obesity on the cytochrome P450 enzyme
system. Int J Clin Pharmacol Ther. 1999;37:8-19.
7. Lewis R. Antifungal drug interactions. Available at:
www.doctorfungus.org/thedrugs/antif_interaction.htm. Accessed January 12, 2007.
8. Clark CM, et al. Methotrexate and cyclosporine for severe recalcitrant
psoriasis. British J Dermatol. 1999;141:279-282.
9. Rheumatrex (methotrexate) package insert. STADA Pharmaceuticals, Cranbury,
NJ 2003.
10. Bloom EJ, et al. Delayed clearance of methotrexate associated with
antibiotics and anti-inflammatory agents. Clin Res. 1986;34;560A.
11. Ronchera CL, et al. Pharmacokinetic interaction between high dose
methotrexate and amoxicillin. Ther Drug Monit. 1993;15:375-379.
12. Tracy TS, et al. The effects of a salicylate, ibuprofen, and naproxen on
the disposition of methotrexate in patients with rheumatoid arthritis. Eur
J Clin Pharmacol. 1992;42:121-125.
13. Tracy TS, et al. Methotrexate disposition following concomitant
administration of ketoprofen, piroxicam and flurbiprofen in patients with
rheumatoid arthritis. British J Clin Pharmacol. 1994;37:453-456.
14. Korstanje MJ, et al. Cyclosporine and methotrexate: a dangerous
combination. J Am Acad Dermatol. 1990;23:320-321.
15. Abdel-Rahman SM, et al. Investigation of terbinafine as a CYP2D6 inhibitor
in vivo. Clin Pharmacol Ther. 1999; 65:465-472.
16. Orap (pimozide) package insert. Gate Pharmaceuticals, Sellersville, PA;
2005.
17. Vloten WA. Pimozide: use in dermatology. Dermatology Online J.
2003;9:3.
18. Flockhart DA, et al. Metabolic interaction between clarithromycin and
pimozide may result in cardiac toxicity. Clin Pharmacol Ther.
1996;59:189A.
19. Desta Z, et al. Effect of clarithromycin on the pharmacokinetics and
pharmacodynamics of pimozide in healthy, poor and extensive metabolizers of
cytochrome P4502D6. Clin Pharmacol Ther. 1999;65:10-20.
20. Venkatakrishnan K, et al. Effects of the antifungal agents on oxidative
drug metabolism. Clin Pharmacokinetic. 2000;38:111-180.
21. Cadle RM, et al. Fluconazole induced symptomatic phenytoin toxicity.
Ann Pharmacother. 1994; 28:292-295.
22. Sporanox package insert. Janssen Pharmaceutical Products, LP, Titusville,
NJ; 2006.
23. Yu, DT, et al. Frequency of potential azole drug-drug interactions and
consequences of potential fluconazole drug interactions.
Pharmacoepidemiology and Drug Safety. 2005;14:755-767.
24. EryC prescribing information. Warner Chilcott, Inc., Rockaway, NJ; 2005.
25. Ciprofloxacin prescribing information. Bayer Pharmaceuticals Corporation,
Westhaven, CT; 2005.
26. Tsankov N, Angelova I. Rifampin in dermatology. Clin Dermatol.
2003;21:50-55.
27. Barranco VP. Update on clinically significant drug interactions in
dermatology. J Am Acad Dermatol. 2006;54:676-684.
To comment on this article, contact editor@uspharmacist.com.



