US Pharm.
2008;33(4):HS-3-HS-21.
Drug-drug interactions are
often a serious complication of taking multiple medications and account for 3%
to 5% of all in-hospital medication errors.1 The consequences of
drug interactions vary, ranging from drug toxicities to therapeutic failures.
These consequences can result in suboptimal treatment of the targeted disease
states, damage to vital organ systems, or death. Drug interactions are of
particular concern in patients infected with HIV who are receiving highly
active antiretroviral therapy (HAART). In a recent retrospective review of
antiretroviral (ARV) drug therapy, logistic regression analysis revealed that
age exceeding 42 years, greater than three comorbidities, and treatment with
three or more ARVs or a protease inhibitor (PI) independently increased the
risk of a clinically significant drug interaction.2 ARVs used in
the treatment of HIV are often prone to drug interactions because many of them
are metabolized through the CYP450 system. Of the CYP450 isoenzymes, CYP3A4,
CYP2D, and CYP2C9/19 are considered the primary isoenzymes involved in the
drugs' metabolism.3
There are six major classes of
HIV ARVs available for use in HAART: nucleoside reverse transcriptase
inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs),
PIs, fusion inhibitors, integrase inhibitors, and chemokine receptor (CCR5)
antagonists. Since NRTIs, fusion inhibitors, and integrase inhibitors do not
undergo hepatic metabolism through the CYP450 system, their drug interaction
profile is minimal. Conversely, NNRTIs, PIs, and maraviroc (a CCR5
antagonist), are extensively metabolized by the CYP450 system; thus, they are
highly susceptible to drug interactions. Furthermore, NNRTIs and PIs can
inhibit and/or induce some of the CYP450 isoforms. For example, PIs are
metabolized by, and are variable inhibitors of, the CYP3A4 isoform. Of the
PIs, ritonavir is the most potent inhibitor, followed by indinavir,
nelfinavir, amprenavir, and saquinavir, in decreasing order.4
Additionally, the PIs are substrates and inhibitors of P-glycoprotein, a major
drug transmembrane efflux protein that transports PIs and other drugs out of
the cell, which can lead to a decrease in the bioavailability of PIs.3
Of the four available NNRTIs, nevirapine and etravirine are substrates, as
well as inducers of, CYP3A4, and delavirdine is a substrate and an inhibitor
of CYP3A4.3,5 Etravirine is an inhibitor of CYP2C9/2C19.5
Efavirenz is considered both an inducer and inhibitor of the CYP3A4 isoform,
with the inducing trait predominating.6 Maraviroc is metabolized by
the CYP3A isoform and is likely to be affected by drugs that either inhibit or
induce this CYP isoform; however, in vitro data indicate that maraviroc itself
has minimal effect on the CYP450 system.7
Overall, the pharmacokinetic
properties of NNRTIs and PIs can lead to sub- or supratherapeutic levels of
concomitant drugs that are metabolized by the same CYP450 isoenzymes.
Alternatively, if the concomitant drug acts as an inhibitor or inducer of the
CYP450 isoenzymes that the NNRTIs, PIs, or maraviroc are metabolized through,
the ARV levels will change accordingly, possibly resulting in a reduced
antivirologic response or ARV-related toxicities to the patient. Understanding
the mechanisms of the CYP450-systemÒmediated drug interactions between ARVs
and commonly prescribed medications in the HIV patient population is important
in order to determine if dosing modifications, close monitoring for adverse
effects of drugs, or careful monitoring of a patient's viral load for
virologic failure are necessary.
This review will discuss major
drug interactions that occur with NNRTIs, PIs, and maraviroc and focus on the
following classes of drugs: antiepileptic drugs (AEDs), statins, methadone,
antidepressants, phosphodiesterase (PDE)-5 inhibitors, and acid-suppressive
agents (TABLE 1).
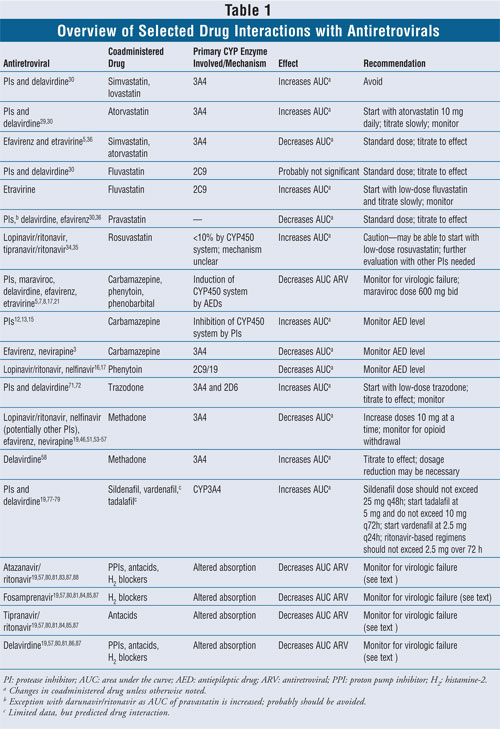
Antiepileptics
The
first-generation AEDs (i.e., carbamazepine, phenytoin, phenobarbital) are
substrates as well as inducers of the CYP450 system.8 Since PIs,
NNRTIs, and maraviroc are metabolized through the same pathways, the potential
exists for significant drug interactions.7,9,10 Several case
reports and reviews have demonstrated that there is a decreased metabolism and
subsequent toxicity of carbamazepine when concomitantly administered with PIs,
particularly ritonavir (including low doses of ritonavir utilized for boosting
other PIs).8,11-15 One study reported an increase in carbamazepine
concentrations from 9.5 mcg/mL to 17.8 mcg/mL within 12 hours of a single dose
of ritonavir 200 mg.12 Therefore, in patients receiving
carbamazepine concurrently with a ritonavir-containing HAART regimen, careful
monitoring of carbamazepine levels is essential. Additionally, it may be
appropriate to initiate a conservative carbamazepine dose in these patients or
choose an alternative AED.12,13,15
Induction of certain CYP450
isoenzymes (i.e., 2C9/19 by lopinavir/ritonavir and nelfinavir) has been
reported and can lead to an increase in the metabolism of AEDs like phenytoin,
a narrow therapeutic index drug. Consequently, this reduction in the
anticonvulsant serum concentrations can lead to seizures.16,17
Likewise, ritonavir-based HAART regimens have been shown to decrease valproic
acid concentrations. One case report demonstrated a 48% decrease in valproic
acid levels after a patient was started on a lopinavir/ritonavir-based regimen.
18 This interaction is likely due to ritonavir's ability to induce
valproic acid metabolism via glucuronidation.18 Both NNRTIs
(efavirenz and nevirapine) have been shown to decrease carbamazepine levels.
The area under the curve (AUC) decreased by 27% when efavirenz was
coadministered with carbamazepine.19,20 This reduction in
carbamazepine levels occurred as a result of the induction of the CYP3A4
isoenzyme by efavirenz and nevirapine.3
Furthermore, carbamazepine,
phenytoin, and phenobarbital all have the potential to induce PI, maraviroc,
delavirdine, efavirenz, and etravirine metabolism and thus reduce serum
concentrations of these drugs, which can lead to ARV resistance and drug
failure.5,7,8,17,21 Recommendations regarding the use of maraviroc
with these AEDs suggest that the dose of maraviroc should be increased to 600
mg twice a day; however, formal recommendations regarding dosing adjustments
of PIs, delavirdine, efavirenz, or etravirine are lacking.5,7
Therefore, careful monitoring for virologic resistance and failure is
essential.
Some of the newer,
second-generation AEDs may have pharmacokinetic advantages over traditional
agents, including decreased drug interactions. However, there are limited
clinical data to support their use in combination with ARVs. Gabapentin,
levetiracetam, and lamotrigine are minimally metabolized by the CYP450 system
and may be considered when the potential for significant drug interactions
limits the use of first-generation AEDs.8,22 Use of other
second-generation AEDs (e.g., topiramate, tiagabine, oxcarbazepine, felbamate)
may be limited due to their induction/inhibition of CYP450 isoenzymes and/or
their adverse-effect profiles.8
Due to the difficulty in
predicting the potential interactions between first-generation AEDs and ARVs,
strict monitoring of serum concentrations of these AEDs are necessary in order
to avoid drug toxicity or inadequate control of seizures/disease states.
Dosing adjustments should be made according to serum AED concentrations,
control of the patient's disease state, and adverse effects. Moreover, it is
important to also monitor the patient's virologic response to HAART since some
AEDs have the potential to accelerate the clearance of some ARVs, resulting in
a likely decrease in efficacy.8,15,17,23,24 Alternatively, other
second-generation AEDs that are minimally metabolized by the CYP450 system may
be suitable alternatives if deemed appropriate for the patient.8,22
Statins
Although HAART has
decreased the morbidity and mortality rates among HIV patients, these drugs
have also been associated with long-term metabolic complications (e.g.,
lipodystrophy, dyslipidemia, diabetes mellitus).25,26 The PIs in
particular have been associated with causing dyslipidemia, with a prevalence
as high as 74%.27 In addition, dyslipidemia can also be caused by
HIV infection itself.28 Similarly to non-HIV-infected individuals,
HIV patients will often require treatment of their dyslipidemia with
lipid-lowering therapy such as HMG-CoA reductase inhibitors (statins),
fibrates, niacin, or ezetimibe.29 Many statins are metabolized by
the CYP450 system, specifically the CYP3A4 isoenzyme.30 Exceptions
to this are pravastatin, fluvastatin, and rosuvastatin, which are eliminated
through varying pathways.31,32
Since all of the PIs and
delavirdine inhibit CYP3A4, drug interactions with statins are inevitable for
those metabolized by the same enzyme. This interaction results in increased
levels of statins, which could increase the patient's risk for adverse effects
such as myalgias, rhabdomyolysis, elevated creatinine phosphokinase (CPK), and
hepatic dysfunction.29,31 One study demonstrated that the 24-hour
area under the curve (AUC0-24) for simvastatin increased by more
than 3,000% when it was coadministered with saquinavir/ritonavir.30
Another study demonstrated that the AUC0-24 for atorvastatin
increased more than 800% and the maximum concentration (Cmax)
increased more than 760% when coadministered with tipranavir/ritonavir.33
Two recent studies demonstrated an unexpected drug-drug interaction involving
rosuvastatin and the PIs, lopinavir/ritonavir, and tipranavir/ritonavir.
34,35 When rosuvastatin was concurrently administered with
lopinavir/ritonavir, rosuvastatin's AUC was increased by 2.1-fold and the C
max was increased by 4.7-fold. There were four patients who experienced
elevated CPKs as a result of this increase in rosuvastatin concentrations.
34 Furthermore, the combination of tipranavir/ritonavir and rosuvastatin
10 mg resulted in an increase of 37% in rosuvastatin's AUC, and the Cmax
was increased by 123%.35 The mechanisms of these drugÒdrug
interactions are currently unclear.34,35
Interactions with the NNRTIs
have also been established. In one study, volunteers receiving concomitant
efavirenz with either simvastatin, atorvastatin, or pravastatin revealed that
the AUC0-24 decreased by 58%, 43%, and 40%, respectively.36
This study suggests that patients taking a drug considered to be an inducer
of the CYP450 system will probably require higher doses of interacting statins
in order to achieve optimal lipid-lowering effects.36 Since
etravirine is also an inducer of CYP450 3A4, atorvastatin, simvastatin, and
lovastatin concentrations will likely be decreased when coadministered with
etravirine. Additionally, since fluvastatin is metabolized by CYP2C9 and
etravirine is an inhibitor of this isoenzyme, fluvastatin concentrations will
be increased when it is taken concurrently with etravirine.5
Presently, the statins
recommended for use in HIV patients on a PI-based regimen are atorvastatin,
fluvastatin, and pravastatin.29,37-39 Both simvastatin and
lovastatin should not be used with a PI or delavirdine-containing HAART
regimen due to elevations in serum concentrations of the statin.28,38-42
The concurrent use of rosuvastatin and PIs should be used with caution until
additional studies have been performed with other PIs besides
lopinavir/ritonavir and tipranavir/ritonavir. When initiating statin therapy
in an HIV patient, it is advisable to start with the lowest dose available and
titrate up gradually in order to achieve the necessary lipid-lowering effect.
Another option may be to consider alternative lipid-lowering drugs such as
fibrates, ezetimibe, or niacin. All of these bypass the hepatic P450 system
and undergo different routes of metabolism, thus eliminating the potential for
major drug interactions with ARVs.29,40-44
Methadone
Methadone, commonly
prescribed for HIV patients for the treatment of pain or drug addiction,
interacts with most PIs and NNRTIs through several complex mechanisms. These
include induction of the CYP450 system and glucuronyltransferase, changes in
plasma protein binding, induction of P-glycoprotein, and unequal
stereoselective metabolism of the active R-isomer and inactive S-isomer of
methadone.45-47 Methadone is metabolized primarily by CYP3A4 and,
to a lesser degree, by CYP2D6, 2B6, and 1A2 isoenyzmes.45,48-50
Numerous studies have demonstrated that when most PIs, efavirenz, and
nevirapine are administered concurrently with methadone, an unexpected
decrease in methadone concentration results, which can potentially lead to
opiate withdrawal symptoms; however, there seems to be a discordance between
the degree of decrease in methadone concentrations and the risk of opioid
withdrawal.45,46,51,52 It has been hypothesized that this
discordance may be due to an increase in the metabolism of the inactive
S-isomer of methadone or to alterations in methadone protein binding.45
Of the PIs, this drug-drug
interaction with methadone is more pronounced with lopinavir/ritonavir and
nelfinavir, with decreases in methadone AUCs of 53% and 47%, respectively.
53,54 Efavirenz and nevirapine, both NNRTIs and known inducers of
CYP3A4, have also been shown to cause significant decreases in methadone
concentrations when coadministered.55 Concentrations of methadone
were reduced up to 60% when taken with efavirenz and by 46% with nevirapine.
19,46,56,57 Managing these drug interactions can be challenging due to
the unpredictable changes in serum methadone concentrations and correspondence
to patient response; however, changes in methadone doses should be guided by
patient opioid response and signs and symptoms of opioid withdrawal. If
methadone doses need to be increased, it is recommended that the doses be
augmented 10 mg at a time and all changes be accompanied by careful monitoring.
51 If opioid withdrawal symptoms are present, a dose increase of 20 mg
may also be acceptable.51
The NNRTI delavirdine is a
CYP3A4 inhibitor, and coadministration of this medication results in increased
methadone concentrations. Based on a pharmacokinetic study, the AUC was
increased by 19%.58 When methadone therapy is needed in conjunction
to a delavirdine-containing HAART regimen, patient response to methadone
should be closely monitored, and it may be necessary to decrease the dose of
methadone.58
Antidepressants
Depression is a
frequent disorder among HIV patients, with a reported incidence of up to 47.8%
in some studies.59,60 Patients are often treated with a variety of
antidepressants, including selective serotonin reuptake inhibitors (SSRIs).
The majority of ARVs (PIs and NNRTIs) and antidepressants are substrates for,
and can inhibit or induce, the CYP450 system, and they have the potential to
cause clinically significant drug interactions including serotonin syndrome, a
potentially fatal complication.61-63 Most of these drugÒdrug
interactions are mediated through CYP450 2D6 and 3A4 isoenzymes.61
Ritonavir has been shown to
augment levels of a variety of SSRIs, including fluoxetine, citalopram,
paroxetine, and sertraline.63,64 One study reported cases of
serotonin syndrome in patients taking concurrent fluoxetine with either
ritonavir-, saquinavir/ritonavir-, or efavirenz-based regimens.63
These cases were managed by either decreasing the daily dose of fluoxetine by
half or discontinuing ritonavir. Monitoring for antidepressant side effects
when initiating ritonavir in patients receiving concurrent SSRI therapy is
recommended.65 Other ARVs are anticipated to affect SSRIs to a
lesser extent, and initial dose adjustments may not be necessary.61
Fluvoxamine, a potent
inhibitor of CYP1A2 along with fluoxetine and paroxetine (both potent
inhibitors of CYP2D6), may also cause PI toxicity by increasing their
concentrations.61,64,66,67 A statistically significant increase of
19% in the AUC for ritonavir was seen with concomitant fluoxetine
administration.67 However, considering the magnitude of the change,
no ritonavir dose adjustment was recommended. Sertraline, citalopram, and
escitalopram appear to have little effect on the major CYP isoforms and are
not expected to affect levels of the ARVs.66
Alternatives to SSRIs,
including bupropion, mirtazapine, nefazodone, and venlafaxine, have also been
shown to be effective in treating depression in HIV-infected patients.
65,66,68,69 These drugs, together with trazodone, are metabolized by the
CYP isoenzymes, leaving the potential for interactions with PIs (especially
ritonavir) and NNRTIs.61,70 In a blinded study, ritonavir decreased
the clearance of trazodone (metabolized by CYP3A4 and 2D6), resulting in
increased levels of trazodone, which can cause nausea, dizziness, and
hypotension.71 The AUC for trazodone has been shown to increase
2.4-fold when coadministered with ritonavir.72 However, when
bupropion was coadministered with ritonavir, no adverse effects (i.e.,
seizures) were reported.73 At this time, there are very limited
data on the effects of these alternative antidepressants on the PIs and
NNRTIs. Therefore, when coadministering antidepressants and either a PI- or
NNRTI-based HAART regimen, antidepressants should be initiated at a low dose
and titrated over several weeks. Patients should also be closely monitored for
adverse effects.74
PDE-5 Inhibitors
Erectile
dysfunction is a common occurrence among HIV-infected men, with some studies
demonstrating an incidence as high as 61.4%.75 PDE-5 inhibitors
(e.g., sildenafil, tadalafil, vardenafil) are the cornerstone of therapy for
erectile dysfunction and are considered to be an effective treatment for this
disorder.76
PDE-5 inhibitors are
metabolized through the CYP450 3A4 isoenzyme. Therefore, concentrations of
PDE-5 inhibitors are increased in the presence of PIs and delavirdine.
19,77,78 When saquinavir (the least potent CYP3A4 inhibitor) and
ritonavir (the most potent CYP3A4 inhibitor) were coadministered with
sildenafil, a 3.1-fold and an 11-fold increase in the AUC occurred,
respectively. The Cmax was also increased by 2.4-fold with
saquinavir and 3.9-fold with ritonavir.77 These increases in the
AUC and Cmax of sildenafil suggest that a lower starting dose of
sildenafil should be utilized when initiating therapy in a patient receiving
concurrent therapy with PIs or delavirdine. Limited data exist involving
vardenafil and tadalafil interactions with PIs and delavirdine; however, since
both of these drugs are metabolized primarily through the CYP3A4 isoenzyme, it
can also be expected that the drug concentrations of these PDE-5 inhibitors
will also be increased in the presence of these ARVs.
Potential consequences of
increased concentrations of PDE-5 inhibitors include hypotension, dizziness,
and priapism.79 Therefore, if PDE-5 inhibitor therapy is needed in
a patient receiving a PI- or delavirdine-based HAART regimen, conservative
dosing of these drugs is essential and careful monitoring for adverse effects
of PDE-5 inhibitors is warranted. Recommendations for concurrent therapy with
PIs or delavirdine indicate that sildenafil dosing should not exceed 25 mg
every 48 hours, the starting dose of tadalafil should be 5 mg and not exceed
10 mg within a 72-hour period, and vardenafil should be initiated at 2.5 mg in
a 24-hour period. However, if the HAART regimen contains ritonavir, then the
vardenafil dose should not exceed 2.5 mg within a 72-hour period.19,79
Acid-Suppressive Agents
Acid-suppressive
therapy with histamine-2 (H2) blockers, proton pump inhibitors
(PPIs), or antacids can cause a decrease in the absorption of some PIs and
delavirdine due to changes in the pH of the gastrointestinal tract.80,81
Some PIs (e.g., indinavir) are able to overcome this alteration in absorption
through boosting with ritonavir.82 However, other PIs (e.g.,
atazanavir, fosamprenavir, tipranavir) have been found to have significant
interactions with acid-suppressive therapy that requires intervention due to
the potential for virologic failure from inadequate ARV concentrations.80
Atazanavir/ritonavir's AUC and minimum concentration (Cmin) were
decreased by up to 76% and 71%, respectively, when coadministered with
omeprazole.83 Additionally, fosamprenavir's AUC was decreased by
30% when coadministered with ranitidine (an H2 blocker), and
tipranavir/ritonavir's AUC and Cmin were reduced by 27% and 29%,
correspondingly, when given with antacids.84,85 Of the NNRTIs,
delavirdine can also be affected by antacids, with a 44% reduction in the AUC
when coadministered.86
Recommendations for managing
these interactions vary, but when fosamprenavir, tipranavir/ritonavir, or
delavirdine are used in combination with antacids, it is recommended that the
ARV be given either one to two hours before or one hour after the
administration of the antacid. Fosamprenavir should be boosted with ritonavir
when concurrently administered with H2 blockers.19,57,80,81,87
Atazanavir 300 mg in combination with ritonavir 100 mg should be administered
concurrently with or at least 10 hours after the H2 blocker;
additionally, depending on whether the patient is treatment-naëve or
treatment-experienced, the H2-blocker dose should not surpass 40 mg
or 20 mg twice daily equivalent of famotidine, respectively.88
Atazanavir should not be administered with PPI therapy in individuals who are
treatment-experienced due to the significant reductions in ARV concentrations.
In treatment-naëve patients, however, the PPI dose should not be greater than
20 mg equivalent of omeprazole. Additionally, the PPI must be taken 12 hours
before the atazanavir 300-mg/ritonavir 100-mg dose.88 Data are
lacking in regards to the administration of fosamprenavir or tipranavir with
PPIs, but patients should be closely monitored while receiving concurrent
therapy.19,57,80,81,8
Conclusion
Drug-drug
interactions with HAART, particularly PI- and NNRTI-based regimens, often
complicate the management of patients with HIV. Many of these drug
interactions are mediated through the CYP450 system, particularly the CYP3A4
isoenzyme. PIs, NNRTIs, and maraviroc are metabolized through this same
system; therefore, any drugs that alter the CYP450 system through inhibition
or induction can cause changes in serum concentrations of these ARVs.
Additionally, PIs and NNRTIs can also inhibit or induce CYP450 isoenzymes and
affect concentrations of other drugs that are coadministered with them. As a
clinician, it is imperative to understand these drug interactions in order to
prevent drug toxicities, virologic failure, or death in patients with HIV.
Various resources such as Web sites, databases, and HIV guidelines are
routinely updated and are helpful sources of information that should be
utilized when reviewing HIV patients' medication profiles, especially as this
area continues to expand and more drug interactions are discovered.
REFERENCES
1. Leape LL, Bates DW, Cullen DJ, et al. Systems analysis of adverse drug events. ADE Prevention Study Group. JAMA. 1995;274:35-43.
2. Miller CD, El-Kholi R, Faragon JJ, Lodise TP. Prevalence and risk factors for clinically significant drug interactions with antiretroviral therapy. Pharmacotherapy. 2007;27:1379-1386.
3. de Maat MM, Ekhart GC, Huitema AD, et al. Drug interactions between antiretroviral drugs and comedicated agents. Clin Pharmacokinet. 2003;42:223-282.
4. Hsu A, Granneman GR, Bertz RJ. Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet. 1998;35:275-291.
5. Intelence (etravirine) package insert. Raritan, NJ: Tibotec Therapeutics; January 2008.
6. Maggiolo F. Efavirenz. Expert Opin Pharmacother. 2007;8:1137-1145.
7. Selzentry (maraviroc) package insert. New York, NY: Pfizer; August 2007.
8. Liedtke MD, Lockhart SM, Rathbun RC. Anticonvulsant and antiretroviral interactions. Ann Pharmacother. 2004;38:482-489.
9. Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277:423-431.
10. Mesdjian E, Seree E, Charvet B, et al. Metabolism of carbamazepine by CYP3A6: a model for in vitro drug interactions. Life Sci. 1999;64:827-835.
11. Bates DE, Herman RJ. Carbamazepine toxicity induced by lopinavir/ritonavir and nelfinavir. Ann Pharmacother. 2006;40:1190-1195.
12. Kato Y, Fujii T, Mizoguchi N, et al. Potential interaction between ritonavir and carbamazepine. Pharmacotherapy. 2000;20:851-854.
13. Burman W, Orr L. Carbamazepine toxicity after starting combination antiretroviral therapy including ritonavir and efavirenz. AIDS. 2000;14:2793-2794.
14. Mateu-de AJ, Grau S, Gimeno-Bayon JL, Carmona A. Ritonavir-induced carbamazepine toxicity. Ann Pharmacother. 2001;35:125-126.
15. Garcia AB, Ibarra AL, Etessam JP, et al. Protease inhibitor-induced carbamazepine toxicity. Clin Neuropharmacol. 2000;23:216-218.
16. Honda M, Yasuoka A, Aoki M, Oka S. A generalized seizure following initiation of nelfinavir in a patient with HIV type 1, suspected due to interaction between nelfinavir and phenytoin. Intern Med. 1999;38:302-303.
17. Lim ML, Min SS, Eron JJ, et al. Coadministration of lopinavir/ritonavir and phenytoin results in two-way drug interaction through cytochrome P-450 induction. J Acquir Immune Defic Syndr. 2004;36:1034-1040.
18. Sheehan NL, Brouillette MJ, Delisle MS, Allan J. Possible interaction between lopinavir/ritonavir and valproic acid exacerbates bipolar disorder. Ann Pharmacother. 2006;40:147-150.
19. Panel on Antiretroviral
Guidelines for Adults and Adolescents. Guidelines for the use of
antiretroviral agents in HIV-infected adults and adolescents. HHS. January 29,
2008. www.aidsinfo.nih.gov/ContentFiles/
AdultandAdolescentGL.pdf. Accessed February 15, 2008.
20. Sustiva (efavirenz) package insert. Princeton, NJ: Bristol-Myers Squibb; January 2007.
21. Hugen PW, Burger DM, Brinkman K, et al. Carbamazepineñindinavir interaction causes antiretroviral therapy failure. Ann Pharmacother. 2000;34:465-470.
22. Nicolas JM, Collart P, Gerin B, et al. In vitro evaluation of potential drug interactions with levetiracetam. Drug Metab Dispos. 1999;27:250-254.
23. Ferrando SJ, Wapenyi K. Psychopharmacological treatment of patients with HIV and AIDS. Psychiatr Q. 2002;73:33-49.
24. Romanelli F, Pomeroy C. Concurrent use of antiretrovirals and anticonvulsants in HIV seropositive patients. Curr Pharm Des. 2003;9:1433-1439.
25. Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12:F51-F58.
26. Geletko SM, ZuWallack AR. Treatment of hyperlipidemia in HIV-infected patients. Am J Health Syst Pharm. 2001;58:607-614.
27. Carr A, Samaras K, Thorisdottir A, et al. Diagnosis, prediction, and natural course of HIV-1 protease-inhibitor-associated lipodystrophy, hyperlipidaemia, and diabetes mellitus: a cohort study. Lancet. 1999;353:2093-2099.
28. Caramelli B, de Bernoche CY, Sartori AM, et al. Hyperlipidemia related to the use of HIV-protease inhibitors: natural history and results of treatment with fenofibrate. Braz J Infect Dis. 2001;5:332-338.
29. Dube MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in HIV-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the IDSA and the Adult AIDS Clinical Trials Group. Clin Infect Dis. 2003;37:613-627.
30. Fichtenbaum CJ, Gerber JG, Rosenkranz SL, et al. Pharmacokinetic interactions between protease inhibitors and statins in HIV seronegative volunteers: ACTG Study A5047. AIDS. 2002;16:569-577.
31. Davidson MH, Toth PP. Comparative effects of lipid-lowering therapies. Prog Cardiovasc Dis. 2004;47:73-104.
32. Everett DW, Chando TJ, Didonato GC, et al. Biotransformation of pravastatin sodium in humans. Drug Metab Dispos. 1991;19:740-748.
33. Aptivus (tipranavir) package insert. Ridgefield, CT: Boehringer Ingelheim; October 2007.
34. Hoody D, Kiser J, Predhomme J, et al. Drug-drug interaction between lopinavir/ritonavir and rosuvastatin. 14th Conference on Retroviruses and Opportunistic Infections; February 25-28, 2007; Los Angeles, CA.
35. Pham P, Lee L, Fuchs E, et al. Pharmacokinetic interaction between tipranavir/ritonavir and rosuvastatin. 15th Conference on Retroviruses and Opportunistic Infections; February 3-6, 2008; Boston, MA.
36. Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr. 2005;39:307-312.
37. Moyle GJ, Lloyd M, Reynolds B, et al. Dietary advice with or without pravastatin for the management of hypercholesterolaemia associated with protease inhibitor therapy. AIDS. 2001;15:1503-1508.
38. Henry K, Melroe H, Huebesch J, et al. Atorva!=statin and gemfibrozil for protease-inhibitor-related lipid abnormalities. Lancet. 1998;352:1031-1032.
39. Doser N, Kubli S, Telenti A, et al. Efficacy and safety of fluvastatin in hyperlipidemic PI-treated HIV-infected patients. AIDS. 2002;16:1982-1983.
40. Niaspan (niacin) package insert. North Chicago, IL: Abbott Laboratories; September 2007.
41. Fessel WJ, Follansbee SE, Rego J. High-density lipoprotein cholesterol is low in HIV-infected patients with lipodystrophic fat expansions. AIDS. 2002;16:1785-1789.
42. Palacios R, Santos J, Gonzalez M, Ruiz J, et al. Efficacy and safety of fenofibrate for the treatment of hypertriglyceridemia associated with antiretroviral therapy. J Acquir Immune Defic Syndr. 2002;31:251-253.
43. Miller J, Brown D, Amin J, et al. A randomized, double-blind study of gemfibrozil for the treatment of protease inhibitor-associated hypertriglyceridaemia. AIDS. 2002;16:2195-2200.
44. Bennett MT, Johns KW, Bondy GP. Ezetimibe is effective when added to maximally tolerated lipid lowering therapy in patients with HIV. Lipids Health Dis. 2007;6:15.
45. Gerber JG. Using pharmacokinetics to optimize antiretroviral drug-drug interactions in the treatment of HIV. Clin Infect Dis. 2000;30(suppl 2):S123-S129.
46. Clarke S, Mulcahy F, Bergin C, et al. Absence of opioid withdrawal symptoms in patients receiving methadone and the protease inhibitor lopinavir-ritonavir. Clin Infect Dis. 2002;34:1143-1145.
47. Barry M, Feely J. Enzyme induction and inhibition. Pharmacol Ther. 1990;48:71-94.
48. Iribarne C, Berthou F, Baird S, et al. Involvement of cytochrome P450 3A4 enzyme in the N-demethylation of methadone in human liver microsomes. Chem Res Toxicol. 1996;9:365-673.
49. Wu D, Otton SV, Sproule BA, et al. Inhibition of human cytochrome P450 2D6 (CYP2D6) by methadone. Br J Clin Pharmacol. 1993;35:30-34.
50. Felder C, Uehlinger C, Baumann P, et al. Oral and IV methadone use: some clinical and pharmacokinetic aspects. Drug Alcohol Depend.1999;55:137-143.
51. Gourevitch MN, Friedland GH. Interactions between methadone and medications used to treat HIV infection. Mt Sinai J Med. 2000;67:429-436.
52. Stevens RC, Rapaport S, Maroldo-Connelly L, et al. Lack of methadone dose alterations or withdrawal symptoms during therapy with lopinavir/ritonavir. J Acquir Immune Defic Syndr. 2003;33:650-651.
53. Kaletra (lopinavir/ritonavir) package insert. North Chicago, IL: Abbott Laboratories; November 2007.
54. Smith PF, Booker BM, Difrancesco R, et al. Effect of methadone or LAAM on the pharmacokinetics of nelfinavir and M8. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; December 16-19, 2001; Chicago, IL.
55. Pinzani V, Faucherre V, Peyriere H, Blayac JP. Methadone withdrawal symptoms with nevirapine and efavirenz. Ann Pharmacother. 2000;34:405-407.
56. Calvo R, Lukas JC, Rodriguez M, et al. Pharmacokinetics of methadone in HIV-positive patients receiving the non-nucleoside reverse transcriptase efavirenz. Br J Clin Pharmacol. 2002;53:212-214.
57. Back DJ, Owen A, Khoo SH. Population pharmacokinetics of efavirenz in an unselected cohort of HIV-1-infected individuals. Clin Pharmacokinet. 2006;45:213-214.
58. McCance-Katz EF, Rainey PM, Smith P, et al. Drug interactions between opioids and antiretroviral medications: interaction between methadone, LAAM, and delavirdine. Am J Addict. 2006;15:23-34.
59. Dew MA, Becker JT, Sanchez J, et al. Prevalence and predictors of depressive, anxiety and substance use disorders in HIV-infected and uninfected men: a longitudinal evaluation. Psychol Med. 1997;27:395-409.
60. Ciesla JA, Roberts JE. Meta-analysis of the relationship between HIV infection and risk for depressive disorders. Am J Psychiatry. 2001;158:725-730.
61. Tseng AL, Foisy MM. Significant interactions with new antiretrovirals and psychotropic drugs. Ann Pharmacother. 1999;33:461-743.
62. Baker GB, Fang J, Sinha S, Coutts RT. Metabolic drug interactions with SSRI antidepressants. Neurosci Biobehav Rev. 1998;22:325-333.
63. DeSilva KE, Le Flore DB, Marston BJ, Rimland D. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS. 2001;15:1281-1285.
64. Angelino AF, Treisman GJ. Management of psychiatric disorders in patients infected with HIV. Clin Infect Dis. 2001;33:847-856.
65. Penzak SR, Reddy YS, Grimsley SR. Depression in patients with HIV infection. Am J Health Syst Pharm. 2000;57:376-386.
66. Thompson A, Silverman B, Dzeng L, Treisman G. Psychotropic medications and HIV. Clin Infect Dis. 2006;42:1305-1310.
67. Ouellet D, Hsu A, Qian J, et al. Effect of fluoxetine on pharmacokinetics of ritonavir. Antimicrob Agents Chemother. 1998;42:3107-3112.
68. Currier MB, Molina G, Kato M. A prospective trial of sustained-release bupropion for depression in HIV-seropositive and AIDS patients. Psychosomatics. 2003;44:120-125.
69. Elliott AJ, Russo J, Bergam K, et al. Antidepressant efficacy in HIV-seropositive outpatients with major depressive disorder: an open trial of nefazodone. J Clin Psychiatry. 1999;60:226-231.
70. Hesse LM, von Moltke LL, Shader RI, et al. Ritonavir, efavirenz, and nelfinavir inhibit CYP2B6 activity in vitro: potential drug interactions with bupropion. Drug Metab Dispos. 2001;29:100-102.
71. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Short-term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol. 2003;43:414-422.
72. Norvir (ritonavir) package insert. North Chicago, IL: Abbott Laboratories; May 2007.
73. Park-Wyllie LY, Antoniou T. Concurrent use of bupropion with CYP2B6 inhibitors, nelfinavir, ritonavir and efavirenz. AIDS. 2003;17:638-640.
74. Caballero J, Nahata MC. Use of SSRIs in the treatment of depression in adults with HIV. Ann Pharmacother. 2005;39:141-145.
75. Crum-Cianflone NF, Bavaro M, Hale B, et al. Erectile dysfunction and hypogonadism among men with HIV. A IDS Patient Care STDS. 2007;21:9-19.
76. Montague DK, Jarow JP, Broderick GA, et al. Chapter 1: The management of erectile dysfunction: an AUA update. J Urol. 2005;174:230-239.
77. Muirhead GJ, Wulff MB, Fielding A, et al. Pharmacokinetic interactions between sildenafil and saquin!=!=avir/ritonavir. Br J Clin Pharmacol. 2000;50:99-107.
78. Scott LJ, Perry CM. Delavirdine: a review of its use in HIV infection. Drugs. 2000;60:1411-1444.
79. Merry C, Barry MG, Ryan M, et al. Interaction of sildenafil and indinavir when co-administered to HIV-positive patients. AIDS. 1999;13:F101-F107.
80. Fulco PP, Vora UB, Bearman GM. Acid suppressive therapy and the effects on protease inhibitors. Ann Pharmacother. 2006;40:1974-1983.
81. Tran JQ, Gerber JG, Kerr BM. Delavirdine: clinical pharmacokinetics and drug interactions. Clin Pharmacokinet. 2001;40:207-226.
82. Rublein JC, Donovan BJ, Hollowell SB, et al. Effect of omeprazole on plasma concentrations of indinavir in HIV-negative subjects. 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy; September 14-17, 2003; Chicago, IL.
83. Agarwala S, Gray K, Wang Y, Grasela D. Pharmacokinetic effect of omeprazole on atazanavir with ritonavir in healthy subjects. 12th Conference on Retroviruses and Opportunistic Infections; February 22-25, 2005; Boston, MA.
84. Ford SL, Wire MB, Lou Y, et al. Effect of antacids and ranitidine on the single-dose pharmacokinetics of fosamprenavir. Antimicrob Agents Chemother. 2005;49:467-469.
85. Van Heeswijk R, Sabo J, Cooper C, et al. The pharmacokinetic interactions between tipranavir/ritonavir 500/200 mg bid (TPV/r) and atorvastatin, antacid and CYP3A4 in healthy adult volunteers. 5th International Workshop on Clinical Pharmacology of HIV Therapy; April 1-3, 2004; Rome, Italy.
86. Rescriptor (delavirdine) package insert. New York, NY: Pfizer; June 2006.
87. Kandula VR, Khanlou H, Farthing C. Tipranavir: a novel second-generation nonpeptidic protease inhibitor. Expert Rev Anti Infect Ther. 2005;3:9-21.
88. Reyataz (atazanavir) package
insert. Princeton, NJ: Bristol-Myers Squibb; December 2007.
To comment on this article,
contact
rdavidson@jobson.com.



