US Pharm. 2019;44(12):HS-14-HS-16.
Clostridium difficile infection (CDI) is the most commonly recognized cause of infectious diarrhea in healthcare settings.1 C diff was responsible for almost half a million infections and was associated with approximately 29,000 deaths in 2011.2 Typical patient presentation of CDI includes diarrhea, watery stool(s) or frequent bowel movements for several days, fever, stomach tenderness or pain, loss of appetite, and nausea.3 It is reasonable to expect a patient to be dehydrated from diarrhea and present with complications such as hypotension and confusion.4
For all severities of CDI, oral vancomycin is recommended as one of the first-line treatment options.1 Oral vancomycin is available in two formulations: capsules and solution. Historically, the solution has been compounded using lyophilized vancomycin hydrochloride with sterile water for injection, USP, until the FDA approved FIRVANQ® (vancomycin hydrochloride for oral solution). Vancomycin hydrochloride is poorly absorbed after oral administration. For treatment of CDI, vancomycin acts locally in the lower gastrointestinal (GI) tract, the typical site of infection.5 Therefore, regardless of formulation (dosage form), vancomycin must adequately dissolve or be released prior to reaching the site of action.5
In this study, the dissolution behavior of vancomycin capsules (Vancocin®, ANI Pharmaceuticals), solution prepared from lyophilized vancomycin hydrochloride for injection, USP, and solution prepared from FIRVANQ® was evaluated. Dissolution testing was conducted using pH media mimicking that of the GI tract to support in-vitro bioequivalence of all three vancomycin formulations.
Methods
Physiological relevant dissolution media (pH 1.0, 4.5, and 6.8 buffers) were prepared and degassed per cUSP requirements (vacuum filtration at 41°C) for each dissolution study. 900 mL of media was added to each of the 12 vessels of Hanson SR8 Plus Dissolution Test Station and equilibrated to 37°C ± 0.5°C.
Either a single 250-mg vancomycin capsule (with wire sinker to mitigate buoyancy) or a 10.0-mL aliquot (using a Class A glass pipette) of sample solution (25 mg/mL vancomycin hydrochloride for injection prepared in sterile water for injection or FIRVANQ® reconstituted using documented instructions) equivalent to 250 mg vancomycin was introduced into each of the dissolution vessels. USP Apparatus 2 paddles were operated at 50 revolutions per minute to mix the media at a constant rate.
At the specified sampling times of 5, 10, 15, 20, 30, 45, and 60 minutes, 10-mL aliquots were withdrawn using a syringe and cannula equipped with a 45-μm polyethylene cannula filter (nonadsorption of vancomycin hydrochloride separately evaluated in a filter study for all three dissolution media) and collected. The filtrate was diluted in the respective dissolution media (0.5-mL sample combined with 0.5-mL media) to fall within the analytical range (0.05-0.15 mg/mL vancomycin) of a previously validated, stability-indicating high-performance liquid chromatography (HPLC) method using an Agilent 1200 system with a Phenomenex Gemini, 5-μm, C18, 110A 150 x 4.6 mm column and Empower data acquisition system, under the parameters and conditions stated in the method.
The dissolution samples were analyzed to determine the content of vancomycin dissolved at each time point to generate a release profile to compare the three drug products. System suitability was established using a standard prepared in the corresponding dissolution media. In order to mitigate degradation of vancomycin hydrochloride in solution, the autosampler compartment in the HPLC was chilled to 5°C. For demonstration of specificity, a blank solution of each dissolution media was analyzed to demonstrate noninterference of extraneous peaks at the retention time of vancomycin.
Results
Using a qualified vancomycin working-standard solution; the dissolution data for each sample was processed and results calculated as per the formula provided in the HPLC method. The sample results were then plotted for percentage released versus time in minutes for each drug and dissolution media. The 12 percentage of drug-released profiles obtained for each of three dosage forms are shown in FIGURES 1-3.
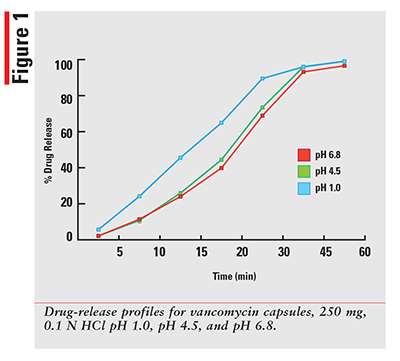
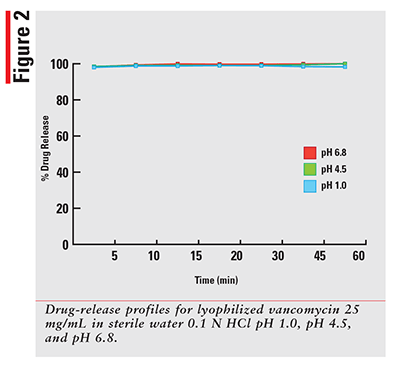
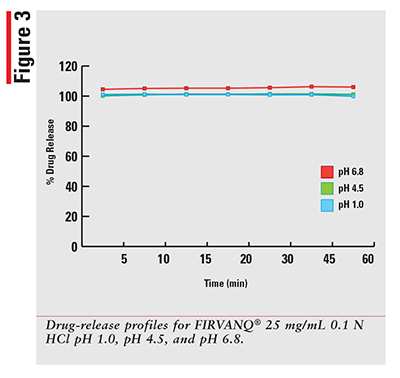
As expected, for a solution that has the drug already dissolved, the observed profiles for FIRVANQ® 25 mg/mL and lyophilized vancomycin hydrochloride for injection 25 mg/mL in sterile water are relatively flat. The study showed complete dissolution of FIRVANQ® 25 mg/mL at the first specified sampling time of 5 minutes. This observation was consistent across all three dissolution media. For vancomycin capsules, a significantly slower dissolution profile is observed. During the 30- to 45-minute interval, less than 80% of the drug was dissolved. Because only the dissolved portion of the drug is available to treat CDI, any drug that is not dissolved is not available for treatment. Using a solution-based delivery formulation such as FIRVANQ® 25 mg/mL, therefore, results in a greater availability in a shorter time frame.
Acknowledgments: All experimental work was performed at a Quality Chemical Laboratories GMP analytical laboratory in Wilmington, North Carolina. Priya Capila, MS, is currently with Sage Therapeutics, Cambridge, Massachusetts.
REFERENCES
1. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):987-994.
2. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825-834.
3. CDC. Clostridioides difficile infection. www.cdc.gov/HAI/organisms/cdiff/Cdiff_infect.html. Accessed February 1, 2019.
4. Manek K, Williams V, Callery S, Daneman N. Reducing the risk of severe complications among patients with Clostridium difficile infection. Can J Gastroenterol. 2011;25(7):368-372.
5. FDA Draft Guidance on Vancomycin Hydrochloride, August 2009.
To comment on this article, contact rdavidson@uspharmacist.com.


