US Pharm.
2006;8:43-51.
Klinefelter's syndrome (KS) is the
constellation of symptoms associated with an extra sex chromosome, the 47, XXY
karotype, compared to the usual male configuration, XY. This group of
symptoms, which include gynecomastia, increased height, sparse facial and body
hair, reduced sperm count, and diminutive testicular size, was originally
observed in nine patients by Dr. Harry Klinefelter in 1942. Many men live with
this abnormality and never become aware of the genetic difference physically
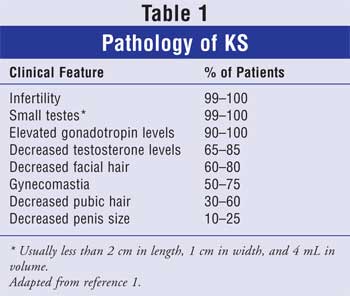
or recognize the associated symptoms (Table 1). Genetic screening for
the chromosomal abnormality began in the 1970s in newborn males via
organizations such as the National Institute of Child Health and Human
Development. KS is not considered an inherited condition. About 25% of the
expected incidence of KS is diagnosed in adulthood. Women who give birth to an
infant with KS do not have a greater risk of recurrence in a later pregnancy
than the risk in the general population.1-3
Genetic Origins
This genetic abnormality occurs
frequently, in 1:500 to 1:1,000 male births. About 3% of the infertile male
population has this condition. The "syndrome" terminology has lost favor in
the literature, since many patients lack clinical evidence of the extra sex
chromosome in youth and may or may not grow to develop symptoms with age. The
nomenclature is now replaced with "XXY males." Research illustrates that half
of the incidence of KS derives from the father's genetic lineage. An increased
risk of KS is linked to advanced maternal age.1
The abnormality is conferred when genetic
material is exchanged during meiosis. This process involves the separation of
one cell (46 chromosomes) into two new cells (23 chromosomes each). During the
exchange between male and female chromosomes, faulty pairing can occur,
leading to an egg with two X chromosomes or to a sperm with an X and Y
chromosome. When a sperm with an XY chromosome fertilizes an egg with a
traditional single X chromosome, or a traditional Y-bearing sperm fertilizes
an egg with two X chromosomes, an XXY male is conceived. A secondary etiology
of this syndrome is an error of parental gametogenesis, when a sperm or egg
carries an extra X chromosome with the normal single-sex chromosome.
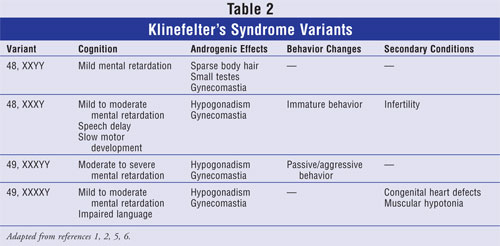
Occasionally, variations of the XXY
abnormality may occur. The most common is the XY mosaic. Some cells are XXY,
and some are XY. While similar outcomes exist between the variations,
fertility is likely improved with the mosaic abnormality (Table 2).2,4
Clinical Symptoms
Primary physical features of XXY
males are gynecomastia (breast enlargement), sparse facial and body hair,
rounded body type due to excess weight, and taller appearance (around 6 feet)
than expected based on paternal and sibling familial history. Approximately
10% of XXY males will seek medical care due to breast enlargement.
Symptoms associated with cognitive and motor
skills include language impairment, which often requires special educational
needs early in childhood development, and delays in toddlers' ability to walk.
Language problems may be seen in school by delays in speech development and
reading and writing abilities. If a child cannot communicate effectively with
single words by 18 to 24 months of age, parents should consult a speech
therapist or language pathologist. Mood can also be negatively affected in
adolescents who develop KS. Teenagers often understand more than they can
articulate in words, compared to their peers. The temperament and disposition
that children display tend to pervade throughout their life.
Testosterone levels are usually low or below
normal, with elevated levels of luteinizing hormone (LH) and
follicle-stimulating hormone (FSH) reaching five to 10 times the normal range
by midpuberty. Testosterone decline is due to failure of the Leydig's cells to
function properly. The pituitary-gonadal system functions effectively through
puberty but begins to change when testosterone falls below normal range,
leading to an overproduction of gonadotropins. The increased production of FSH
and LH leads to hyalinization and fibrosis in the seminiferous tubules, where
the sperm are normally located.
Some clinical features of XXY males are
associated with various health conditions. Incomplete masculinization, a
prominent feature of KS, is linked to infertility, and decreased libido is
associated with osteoporosis. The learning, emotional, and mental disorders of
XXY males are related to taurodontism. Low energy, another clinical feature of
the syndrome, is linked to autoimmune disease.1-7
Diagnosis
Some males may be asymptomatic in
self-reporting based on physical differences or cognitive function. The
likelihood of diagnosis is greatest during birth (before or after), early
childhood, adolescence, or adulthood when testing for infertility. Before
birth, diagnosis can be assessed through amniocentesis or chorionic villus
sampling. These procedures are usually reserved for high-risk pregnancies
associated with familial genetic mutations, pregnant women older than age 35,
or other medical indications that require a sample of amniotic fluid
surrounding the fetus. A definitive diagnosis can be made through a blood
sampling procedure referred to as karotype. White blood cells can be
separated and incubated to detect the extra X chromosome.
XXY males will enter puberty normally
without delay of physical maturity. As puberty progresses, they fail to keep
pace with other males. Normally, testes gradually increase in size from the
initial volume of 2 mL to about 15 mL. In XXY males, while the penis is a
normal size, the testes remain small at 2 mL without a sufficient
quantity of testosterone. To confirm a diagnosis of KS, a thorough medical
history should be conducted to assess developmental abnormalities at birth,
rate and extent of virilization at puberty, sexual functioning, secondary sex
characteristics, beard growth, muscle strength, and energy level.
Endocrine studies primarily illustrate hyper
gonado
tropic hypogonadism secondary to testicular failure. In adult males, the
normal LH is 1.5 to 9 mIU/mL; FSH is 2.0 to 9.2 mIU/mL. In males with KS,
basal serum concentrations of LH and FSH are moderately elevated. Serum
testosterone concentration is usually decreased (<300 ng/dL in adults),
whereas the normal range in adult males is 350 to 1,030 ng/dL. The human
chorionic gonadotropin (hCG) stimulation test typically shows a low to
subnormal testosterone response, with little or no elevation of serum
testosterone concentration after intramuscular (IM) injection of hCG.1-3
Complications
The majority of XXY males do not
produce enough sperm for fertility. XXY males also have a higher risk of
autoimmune diseases (e.g., type 1 diabetes mellitus, lupus erythematosus, and
thyroiditis); this risk is correlated with lower testosterone and higher
estrogen levels. Other hypotheses suggest lymphocyte irregularities. Patients
with hypotestosterone may encounter the onset of osteoporosis later in life,
and bone mineral density has been found to be 12% to 15% lower than normal.
Taurodontism, an enlargement of the pulp of the teeth with surface thinning,
is common in KS and can be diagnosed by dental x-rays. Cerebrovascular
accidents are more prevalent in XXY males, as well as breast cancer,
rheumatoid arthritis, leukemia, and Hodgkin's and non-Hodgkin's lymphoma.
Other systemic, chronic conditions that correlate to KS include lung cancer,
breast cancer, diabetes mellitus, cerebrovascular disease, vascular
insufficiency, nonischemic heart disease, and circulatory diseases.7
Learning disabilities, despite normal or
high IQ, are common. The risk of dyslexia and
attention-deficient/hyperactivity disorder may also be higher. In addition,
psychological problems such as depression are linked to most sexual disorders.
Treatment
Testosterone:
The primary clinical symptoms of KS are treatable. While surgery can correct
gynecomastia, testosterone injections can correct hair loss and promote muscle
mass. Testosterone supplementation should begin in puberty (optimally at age
11 or 12) but is also beneficial in adulthood. Hormonal screening can assess
testosterone levels performed in the morning. The average male produces 4 to 7
mg of testosterone per day in a circadian pattern that peaks in the morning
and is minimal in the evening. However, testicular size, sterility, and
gynecomastia will not be affected by testosterone supplementation.
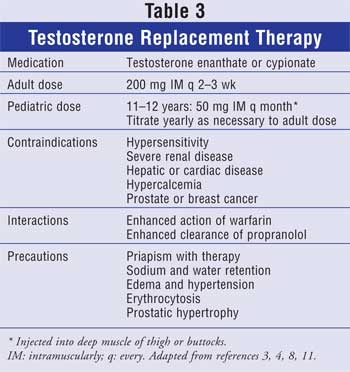
Testosterone can be administered as oral,
buccal, injectable, and transdermal formulations. IM preparations of enanthate
or cypionate are commonly used (Table 3) and have similar
pharmacokinetics and safety profiles. Oral preparations are infrequently
prescribed due to elevations in liver function tests and the risk of
hepatotoxicity. Peak serum levels are achieved in two to five days and return
to baseline approximately two weeks after injection. The typical dose is 200
mg every two weeks. If initiated during puberty, the dose is lowered to 50 to
100 mg every four weeks, then every two weeks until adulthood. Transdermal
testosterone may not be an optimal option due to the scrotal surface area. It
is also a more expensive formulation and is not well studied in patients
younger than 18 years.
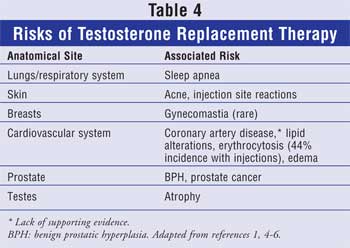
Adverse effects of testosterone use include
allergic reactions (Table 4). The testosterone formulation may be
dissolved in an oil base, causing painful injection site reactions. Weight
gain may also occur from lean body mass and fluid accumulation. Due to
increased androgen accumulation from testosterone, acne may occur. Androgens
can lower high-density lipoprotein levels; thus, close monitoring of lipid
profiles with advancing age is recommended. Continued use of testosterone
beyond age 40 may be linked to benign prostatic hyperplasia. A
prostate-specific antigen and digital rectal exam should be conducted after
three to six months of testosterone therapy in men older than 40 years and
annually thereafter. Nadir testosterone levels should be captured three to
four months prior to the next injection. Levels that exceed 500 ng/dL or that
are less than 200 ng/dL should be dose-adjusted or altered in frequency.
Testosterone is contraindicated in men with breast cancer or known or
suspected prostate cancer. It is also prohibited for use in men with bladder
outlet obstruction, as seen with benign prostatic hypertrophy. Testosterone
supplementation can correct anemia, which is present due to androgen
deficiency from puberty. Hematocrit levels should be monitored, since
elevations above normal range (erythrocytosis) can precipitate an increase in
blood viscosity, aggravating vascular disease in the periphery, heart, and
brain (Table 5).1,2,4,8 Although the etiology is unclear,
testosterone may cause or worsen obstructive sleep apnea.1-4,7 Testosterone
replacement does not mimic normal physiologic sex-steroid production. Thus,
mood fluctuations and physical functioning may be affected.1-4,8

Notably, medications may also contribute to
signs and symptoms of secondary hypogonadism. The patient's medical history
should be screened to avoid exacerbation of testosterone deficiency. These
medications include ketoconazole, glucocorticoids, spironolactone, cimetidine,
phenytoin, opioids, alcohol, and anabolic steroids.
Fertility Treatment:
Advances in fertility treatment, such as sperm extraction, in vitro
fertilization (IVF), and intracytoplasmic sperm insertion (ICSI), can result
in conception without abnormalities or mutations of chromosomes. Additional
options are donor insemination or adoption.
IVF is commonly used for male and female
infertility. Often, the major obstacle for male infertility is the point of
fertilization. IVF is a form of assisted reproductive technology that combines
male sperm with a mature egg in a laboratory dish for fertilization. The
pre-embryo is then transferred to the uterus for implantation.
Through ICSI, a single sperm can be directly
injected into an oocyte (egg). This procedure is usually a second-line option
after IVF has failed, or for males with a severe reduction in sperm count,
which can apply to patients with KS. ICSI does not guarantee pregnancy or
fertilization; it has about a 50% fertilization rate and upward of a 40%
pregnancy rate.9,10
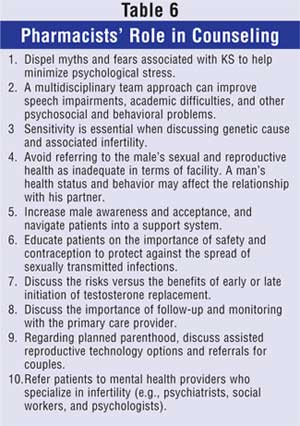
Patient Counseling
Research indicates that the best
time for parents or health care professionals to reveal the presence of KS may
be mid- to late adolescence.1,5,6 At this age, an XXY male is old
enough to understand the condition and may be better equipped to decide whom
he wishes to inform. When educating patients about KS, males should be
reassured that small testes will not interfere with the ability to have a
normal sex life. Patients who develop gynecomastia should be encouraged to
conduct regular breast self-examinations. Concerning testosterone use,
patients can be counseled on painful injection site reactions, since
testosterone may be dissolved in an oil base. Furthermore, pharmacists can
warn patientsthat testosterone may influence mood and physical function.1-4,8
Counseling a patient with KS may prove
challenging to a female pharmacist due to patient sensitivity discussing
sexual and/or fertility difficulties. Female pharmacists may also face unique
challenges in assisting males with KS regarding treatment and education. Table
6 lists counseling tips that address reproductive health concerns, as well
as therapy and fertility options.
The American Association for Klinefelter
Syndrome exists to support research efforts and patient education. Services
include regional support for patients and caregivers, continuing education,
telephone support, referrals, list-serves, and periodic newsletters. The links
listed above can provide patients and parents with useful information and
referral resources.

REFERENCES
1. Smyth CM, Bremner WJ.
Klinefelter syndrome. Arch Intern Med.1998;158:1309-1314.
2. Staessen C, Coonen E, et al.
Preimplantation diagnosis for X and Y normality in embryos from three
Klinefelter patients. Hum Reprod. 1996;11:1650-1653.
3. Matsumoto AM. Hormonal therapy of
male hypogonadism. Endocrinol Metab Clin North Am. 1994;23:857-875.
4. Ghusn HF, Cunningham GR.
Evaluation and treatment of androgen deficiency in males. Endocrinologist.
1991;1:399-405.
5. Smyth CM. Diagnosis and treatment
of Klinefelter syndrome. Hosp Pract. 1999;34:111-120.
6. Staessen C, Tournaye H, et al. PGD
in 47, XXY Klinefelter's syndrome patients. Hum Reprod Update.
2003;9:319-330.
7. Bojesen A, Juul S, et al.
Increased mortality in Klinefelter syndrome. J Clin Endocrinol Metab.
2004;89:3830-3834.
8. Tenover JS. Effects of
testosterone supplementation in the aging male. J Clin Endocrinol Metab.
1992;75:1092-1098.
9. The management of infertility in
teritart care. Royal College of Obstetricians and Gynecologists-Medical
Specialty Society. January 2000.
10. The initial investigation and
management of the infertile couple. Royal College of Obstetricians and
Gynecologists-Medical Specialty Society. Oct. 1998.
11. American Association of Clinical
Endocrinologists and the American College of Endocrinology. AACE Clinical
Practice Guidelines for the Evaluation and Treatment of Hypogonadism in Adult
Male Patients. ED81843; 1196.
To comment on this article, contact
editor@uspharmacist.com.



