US Pharm. 2016;41(1): HS2-HS6.
ABSTRACT: Muscular dystrophy is a genetic, progressive disease with hallmark symptoms of muscle weakness and atrophy. There are nine types of muscular dystrophy, each with varying symptom characteristics and muscle-group involvement. Patients with muscular dystrophy often suffer from cardiac and respiratory complications, as well as impairments in neurologic function. There is currently no known cure for muscular dystrophy. Treatment of the disorder focuses mainly on prevention and management of complications and supportive care. Early diagnosis and appropriate, timely therapy have been demonstrated to prolong life expectancy in patients with muscular dystrophy.
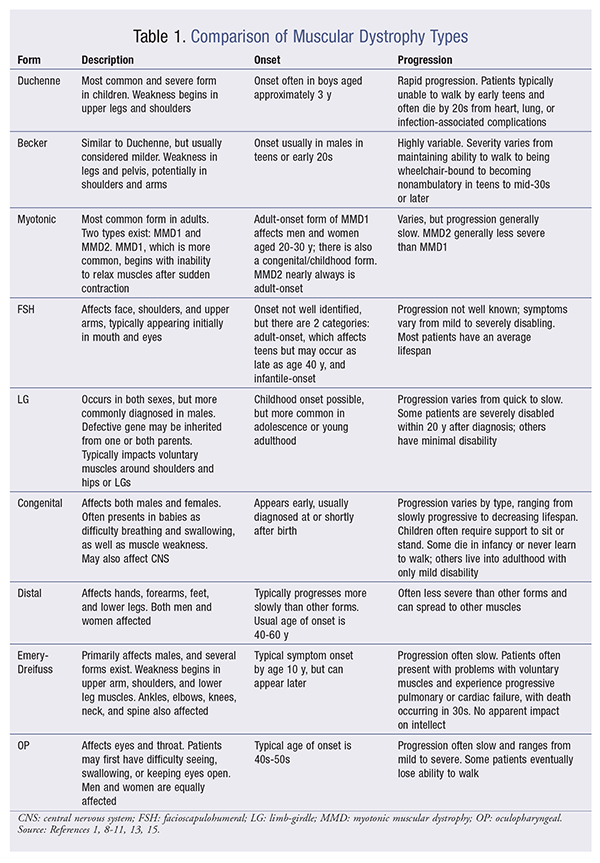
Muscular dystrophy (MD) is a general term used to describe a group of inherited muscle disorders that are progressive in nature, typically leading to weakness and atrophy of varying severity. There are nine clearly defined types of MD, each differing in age of onset, clinical symptomology, muscle groups impacted, and associated complications (TABLE 1). Muscle groups typically affected include the appendicular, axial, and pectoral muscles. Specific forms of the disease may also affect facial, respiratory, and cardiac muscles in addition to tissues or organs such as the brain, eyes, or skin. The disorder may be diagnosed as early as birth or as late as the seventh decade.1
A diagnosis of MD can be devastating for families. Support programs and organizations are available, and family members should be encouraged to use them. The CDC funds the Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet), which collects information about MD in order to improve patient care. The Muscular Dystrophy Association provides grants and training to individuals interested in pursuing research related to genetic muscle disorders.
EPIDEMIOLOGY
Data are lacking concerning the global impact of MD as a whole. The combined prevalence is reported to be between 19.8 and 25.1 cases per 100,000 person-years.2 Duchenne MD (DMD) and Becker MD (BMD) account for more than 80% of all cases, with a reported prevalence of 15.6 per 100,000 boys in Northern England.3,4 DMD, the most common pediatric neuromuscular disease, has an incidence of about 30 per 100,000 or 1 in every 3,500 to 5,000 newborn boys.5-7 The third most common form of MD, facioscapulohumeral MD (FSHD), has an approximate prevalence of 1 per 15,000 to 20,000.8 The prevalence of the congenital forms of MD, as well as dominant and recessive variants, vary by region.1
ETIOLOGY AND PATHOGENESIS
MD is genetically linked, meaning that individuals are born possessing the genes for the disorder even though it may be diagnosed much later in life. The pathogenesis of the MDs is complex and varies depending on disease classification.1 In general, muscle degeneration results from a defective protein whose primary function is critical to the integrity of the cell membrane. When the protein is no longer effective, contents of the cell may leak into the extracellular space, resulting in either necrosis or apoptosis.9-11
The sex-linked forms (dystrophinopathies) include DMD, BMD, and Emery-Dreifuss MD.8 In these X-linked forms of the disease, the defect leads to alterations in the genes coding for the protein dystrophin. Dystrophin, which is widely distributed in smooth, skeletal, and cardiac muscle and in brain tissue, is essential for structural stability of the myofiber. Thus, alterations to the function or availability of this protein leave muscles susceptible to injury and repeated cycles of necrosis and regeneration.10,11
The congenital MDs are autosomal conditions, which in their purest form are due predominately to a total or partial deficiency of the extracellular protein merosin (laminin alpha-2). Although merosin deficiency accounts for 50% of congenital MDs, individual phenotypes are often associated with mutations in different proteins that share similar cellular functions.9,10 The consequence of these mutations is progressive muscle degeneration and, eventually, the loss of independent ambulation.12
Unfortunately, these explanations may not be applicable to all forms of MD, and further research is necessary to better elucidate the disease pathophysiology.13
CLINICAL FEATURES
The hallmark feature of the MDs is weakness of the skeletal muscles, which likely develops as a result of cellular death. The muscles involved—limb, axial, or facial—and the distribution of muscle weakness (proximal/distal) vary according to the type of MD.1 Onset of weakness can become clinically apparent as soon as an infant begins to walk or as late as the second decade of life.8 Ultimately, muscle weakness progresses to loss of ambulation. In addition, patients with MD often develop joint contractures, joint laxity, decreased muscle extensibility, muscle imbalance, decreased bone density (leading to fractures), and spinal deformities such as scoliosis.12
In addition to muscle weakness, patients with MD are susceptible to complications in other organ systems. Dilated cardiomyopathy leading to congestive heart failure, conduction abnormalities, and sudden cardiac death are the most worrisome cardiac ailments. Weakened respiratory muscles and diaphragm, as well as scoliosis, can develop, resulting in respiratory failure. Cognitive impairment, often manifesting as lower IQ scores, mental retardation, attention-deficit/hyperactivity disorder, and autism, may also develop.1 Other common complications of MD include dysphagia, weight loss, pain, hearing loss, and ocular disease.1,8,12,14,15
TREATMENT
To date, there is no definitive cure for any of the MDs. Treatment revolves largely around managing and/or preventing disease complications to improve muscle strength, survival, and quality of life.8,12,15,16
Pharmacologic Treatment
Clinical trials for MD are usually performed in patients with DMD, as it is the most common form. Sample sizes, however, remain small owing to the prevalence of the disease. The most robust data for use of pharmacologic agents in MD involve glucocorticoid therapy.
Prospective, randomized, placebo-controlled trials in DMD patients found that after 6 months of glucocorticoid treatment, patients displayed increased muscle strength, slower progression of weakness, reduced risk of scoliosis, and increased pulmonary function.12,17,18 The optimal dosage in these trials is prednisone 0.75 mg/kg/day, with a maximum dosage of 30 to 40 mg daily. Current guidelines recommend the initiation of glucocorticoids in all patients with DMD once motor function plateaus, with the goal of prolonging ambulation before the decline of motor function.12,19
The decision to treat patients with corticosteroids for longer than 6 months should be individualized with an assessment of risks versus benefits. One retrospective study compared patients treated with glucocorticoids for an average of 8 years with patients who did not receive significant amounts of glucocorticoids.20 Glucocorticoid patients ambulated independently for 3.3 years longer than patients who did not receive glucocorticoid therapy. Additionally, the prevalence of scoliosis was significantly lower in the treatment group. Despite the benefits, patients in the glucocorticoid group had significantly higher rates of long bone and vertebral compression fractures.20 If the decision is made to treat patients with glucocorticoids for a prolonged period of time, adverse effects should be proactively monitored and caregivers should receive appropriate counseling. A dose reduction may be considered, as a daily dosage of prednisone 0.3 mg/kg has been shown effective. Live vaccines should be avoided in these patients or administered before initiation of glucocorticoid therapy.12
Alternative approaches to daily administration of glucocorticoids have been investigated. Compared with daily administration, alternate-day administration resulted in decreased sustainment of muscle strength at 6 months; therefore, alternate-day administration is not recommended.21 A prospective, randomized, double-blind, placebo-controlled equivalence study analyzed weekend versus daily dosing of prednisone. When prednisone was administered for 12 months to steroid-naïve patients, weekend dosing was equivalent to daily dosing for muscle strength, without a significant difference in weight gain.22
A prospective, longitudinal, observational study was performed to determine the long-term benefits of intermittent (10 days on, 10 days off) versus daily glucocorticoids in DMD. Median age for loss of ambulation was 12 years in the intermittent group and 14.5 years in the daily group (statistical significance not reached). The average age for loss of ambulation did not differ between groups. A quicker decline in ambulation after age 7 years was seen in the intermittent group, although declines in respiratory and cardiac function did not differ. Side effects of glucocorticoids occurred in significantly more patients in the daily-administration group than in the intermittent group.23
The benefit of glucocorticoids in other forms of MD is limited. In four patients with BMD, a 6-month trial of prednisolone indicated improvements in muscle strength in some, but not all, muscle groups.24 Furthermore, trials of glucocorticoids in FSHD have shown no benefit.8
Pharmacologic Prevention of Complications
Idebenone, an antioxidant and inhibitor of lipid peroxidation, recently underwent a double-blind, randomized, placebo-controlled phase III trial. To determine the effects of idebenone in patients with DMD, the change in peak expiratory flow at 52 weeks from baseline was analyzed. Compared with placebo, idebenone significantly reduced the loss of respiratory function, with similar adverse effects. Approximately one-half of patients in each group received concomitant glucocorticoid therapy, and nearly all patients were nonambulatory at baseline.25
Given the high prevalence of death from cardiovascular (CV) causes in DMD, mortality improvement and delaying progression of CV diseases have been researched extensively. The effects of perindopril on left ventricular (LV) dysfunction and mortality were evaluated in a randomized, double-blind clinical trial.26,27 Patients with normal LV function were initially assigned to perindopril or placebo for 3 years. After 3 years, both groups received open-label perindopril for the next 7 years. At 5 years, significantly fewer patients originally randomized to perindopril developed an LV ejection fraction <45% compared with the placebo group.26 Furthermore, at the end of 10 years, perindopril patients had a statistically significantly higher survival rate.27
Because of pulmonary compromise late in the disease course, all patients with MD should be appropriately vaccinated against influenza and pneumococcus. To promote increased bone density, calcium and vitamin D should be given. Currently, there is no evidence supporting the prophylactic use of bisphosphonates.12
Pharmacologic Agents in the Pipeline
Two drugs currently under investigation, eteplirsen and drisapersen, have an effect in a small subset of DMD patients whose mutations in the dystrophin gene regain function by skipping exon 51.28-30 Early-stage trials of these two agents have shown a potential benefit in the 6-minute walk test. In addition, gene repair, gene therapy, and stem-cell research are current strategies being considered in the search for a cure.31
Nonpharmacologic Treatment
The cornerstone of therapy for all forms of MD is a comprehensive, collaborative nonpharmacologic treatment plan. Physical therapy, rehabilitation, and stretching/strength exercises are imperative for managing muscle weakness. Frequent orthopedic assessment will determine the patient’s functional status and future interventions. Certain surgical procedures may be performed to relieve scoliosis or contractures.1,16
Not all patients with MD present with typical symptoms of cardiac or respiratory disease; therefore, monitoring is essential. To check for cardiac complications, patients should undergo ECG, echocardiogram, and Holter monitoring. Patients who develop cardiomyopathy or a conduction abnormality should be treated with current accepted medical practices for those disease states. Pulse oximetry and pulmonary-function tests will help identify respiratory insufficiency. Noninvasive ventilation can improve respiratory function.1
Other nonpharmacologic treatment plans include nutritional support and pain control. MD patients may lose the ability to feed themselves as a result of limb-muscle weakness.16 Dysphagia may develop as the disease progresses. Regular assessment of a patient’s nutritional status can prevent malnutrition. Patients with MD experience pain as an expected result of muscle weakness and skeletal abnormalities. Pain should be anticipated, assessed frequently, and treated with acetaminophen, nonsteroidal anti-inflammatory drugs, muscle relaxants, or opioids.10
CONCLUSION
The MDs represent a group of hereditary muscular disorders that lead to muscle weakness and atrophy. Despite the lack of curative treatment for MD, life expectancies have increased in patients owing to the recognition and treatment of complications.12,16 However, most patients succumb to complications of MD, most notably respiratory or CV disease.
REFERENCES
1. Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845-860.
2. Theadom A, Rodrigues M, Roxburgh R, et al. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43:259-268.
3. Mavrogeni S, Markousis-Mavrogenis G, Papavasiliou A, Kolovou G. Cardiac involvement in Duchenne and Becker muscular dystrophy. World J Cardiol. 2015;7:410-414.
4. Norwood FL, Harling C, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132:3175-3186.
5. Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy 14-16th December, 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:682-689.
6. Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48:21-26.
7. Henricson EK, Abresch RT, Cnaan A, et al. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. 2013;48:55-67.
8. Tawil R, Kissel JT, Heatwole C, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;85:357-364.
9. Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur J Hum Genet. 2002;10:91-94.
10. Bushby K. Genetics and the muscular dystrophies. Dev Med Child Neurol. 2000;42:780-784.
11. Miller G, Wessel HB. Diagnosis of dystrophinopathies: review for the clinician. Pediatr Neurol. 1993;9:3-9.
12. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77-93.
13. Emery AE. The muscular dystrophies. Lancet. 2002;359:687-695.
14. Kang PB, Morrison L, Iannaccone ST, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;84:1369-1378.
15. Narayanaswami P, Carter G, David W, et al. Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;84:1720-1721.
16. Bushby K, Bourke J, Bullock R. The multidisciplinary management of Duchenne muscular dystrophy. Curr Paediatr. 2005;15:292-300.
17. Moxley RT III, Ashwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;1:13-20.
18. Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592-1597.
19. Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015;20:18168-18184.
20. King WM, Ruttencutter R, Nagaraja HN, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology. 2007;68:1607-1613.
21. Fenichel GM, Mendell JR, Moxley RT III, et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol. 1991;48:575-579.
22. Escolar DM, Hache LP, Clemens PR, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77:444-452.
23. Ricotti V, Ridout DA, Scott E, et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2013;84:698-705.
24. Bäckman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1995;5:233-241.
25. Buyse GM, Voit T, Schara U, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet. 2015;385:1748-1757.
26. Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855-857.
27. Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154:596-602.
28. Flanigan KM, Voit T, Rosales XQ, et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trial. Neuromuscul Disord. 2014;24:16-24.
29. Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637-647.
30. Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014;13:987-996.
31. Ruegg UT. Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr Opin Neurol. 2013;26:577-584.
To comment on this article, contact rdavidson@uspharmacist.com.



