US Pharm. 2009;34(10):37-47.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2008-2009 (TABLE) detail the basic clinical and pharmacologic profile for each new drug and provide a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience has demonstrated that many aspects of a new drug's therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have clearly shown the appearance of "new" adverse reactions for many NMEs within 2 to 3 years of becoming available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or be withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners become aware of changes in a drug's therapeutic profile as reported by their own patients and in the pharmaceutical literature.

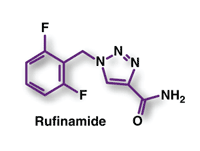
Rufinamide (Banzel, Eisai)
Indication and Clinical Profile1,2: Rufinamide, a novel triazole antiepileptic drug (AED), has been approved by the FDA for adjunctive treatment of seizures associated with Lennox-Gastaut syndrome (LGS) in patients aged ≥4 years. LGS is a severe form of childhood-onset epilepsy characterized by multiple types of seizures that are typically refractory to treatment; mental retardation frequently accompanies the disorder. Valproic acid, although not approved for use, is considered first-line treatment for LGS. Besides rufinamide, other adjunctive agents approved for LGS treatment include topiramate, lamotrigine, felbamate, and clonazepam.
Approval of rufinamide was based on a randomized, multicenter, double-blind, placebo-controlled, 12-week, add-on trial in 138 highly refractory LGS patients (≥90 seizures with ≤3 AEDs in month before baseline) aged 4 to 30 years. In patients treated with rufinamide, the median number of seizures per 28 days decreased significantly from 290 (baseline) to 204. Tonic-atonic seizures per 28 days decreased from 92.0 to 60.7 in rufinamide-treated patients compared with 92.5 to 76.2 in placebo-treated patients. At the trial's end, seizure severity had decreased in 39 of 73 patients treated with rufinamide and in 19 of 62 taking placebo. Status epilepticus occurred in 3 of 74 patients taking rufinamide versus 0 of 64 placebo-treated patients. Rufinamide also has been shown to reduce the frequency of partial seizures, but the drug is not approved for this indication.
Pharmacology and Pharmacokinetics1,2: The precise mechanism by which rufinamide exerts its antiepileptic effect is unknown. In vitro studies suggest that the principal mechanism may be the modulation of central nervous system sodium channel activity--in particular, prolongation of the inactive state of the channel.
Rufinamide is well absorbed after oral administration, but the rate of absorption is relatively slow and the extent is decreased with increased dose. Pharmacokinetics do not change with multiple dosing. Rufinamide is extensively metabolized by carboxylesterases with no active metabolites. Renal excretion is the predominant route of elimination (85%), and the drug's plasma half-life is approximately 6 to 10 hours.
Adverse Reactions, Precautions, and Warnings1,2: In clinical trials, somnolence, fatigue, vomiting, headache, dizziness, nausea, diplopia, and tremor occurred with rufinamide at a higher frequency than with placebo. All AEDs increase the risk of suicidal thoughts or behavior; patients therefore should be monitored for emergence or worsening of depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. Shortening of the QT interval (>20 msec) occurred in 46% of patients taking rufinamide 2,400 mg or 3,200 mg and in 65% of those taking 4,800 mg. While the observed degree of QT shortening was never <300 msec and is without any known clinical risk, rufinamide is contraindicated in patients with familial short QT syndrome. Caution is advised if rufinamide is used with other drugs that shorten the QT interval, (e.g., digoxin, lamotrigine, magnesium). Rufinamide is classified as pregnancy category C.
Drug Interactions1,2: Serum concentrations of rufinamide can be affected by concurrent use of some other AEDs. Valproate has been shown to increase rufinamide plasma levels by up to 70% in children. Rufinamide concentrations decrease 20% to 45% with coadministration of cytochrome inducers, including phenytoin, primidone, phenobarbital, and carbamazepine. Rufinamide does not appear to significantly alter plasma levels of other AEDs, with the exception of a 21% increase in phenytoin levels. Rufinamide appears to be a weak inducer of CYP3A4 and has been shown to reduce ethinyl estradiol (EE), norethindrone, and triazolam exposure. The clinical significance of these interactions has not been established, but reduced efficacy of these and other drugs metabolized by CYP3A4 is possible.
Dosage and Administration1,2: Rufinamide is supplied as 200-mg and 400-mg film-coated, scored tablets. Tablets may be administered whole, in half, or crushed, and should be taken with food. In children aged ≥4 years, treatment should be initiated at approximately 10 mg/kg/day. The dose should be increased by approximately 10-mg/kg increments every other day to a target of 45 mg/kg/day or 3,200 mg/day, whichever is less. In adults, treatment should be initiated at 400 mg to 800 mg/day and increased by 400 mg to 800 mg/day every 2 days until a maximum of 3,200 mg/day is reached. It is not known whether doses <3,200 mg are effective. Rufinamide should always be administered in two equally divided doses. Caution should be exercised in patients with mild-to-moderate hepatic impairment, and use in severe hepatic impairment is not recommended. Patients taking valproate should be started on a lower dose of rufinamide (<400 mg/day).
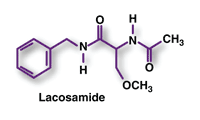
Lacosamide (Vimpat, UCB)
Indication and Clinical Profile3,4: Epilepsy is a chronic neurologic disorder affecting approximately 50 million people worldwide and 3 million people in the U.S. Fewer than half of patients will attain seizure control with their first AED, and more than 30% will continue to experience seizures despite trying two or more AEDs. Lacosamide is a novel AED approved as add-on therapy for the treatment of partial-onset seizures in patients aged ≥17 years with epilepsy.
FDA approval of lacosamide was based on results of three 12-week, randomized, double-blind, placebo-controlled, multicenter trials involving 1,300 subjects with partial-onset seizures who were not adequately controlled with one to three concomitant AEDs. A statistically significant effect was observed for lacosamide at doses of 200 mg/day (Study 3), 400 mg/day (Studies 1, 2, and 3), and 600 mg/day (Studies 1 and 2). Lacosamide patients had a 50% reduction in seizures and experienced reductions in median seizure frequency at significantly greater rates compared with placebo patients.
Pharmacology and Pharmacokinetics3,4: The mechanism by which lacosamide exerts its antiepileptic effect in humans is not fully understood. In vitro studies have shown that lacosamide selectively enhances slow inactivation of voltage-gated sodium channels, resulting in stabilization of hyperexcitable neuronal membranes and inhibition of repetitive neuronal firing. Modulation of sodium channels may reduce the sodium-channel overactivity associated with seizure initiation. This action is different from the biochemical actions of other sodium channel-blocking drugs.
Lacosamide is completely absorbed (100% bioavailability) after oral administration, with negligible first-pass effect. It is a CYP2C19 substrate; the major metabolite formed is O-desmethyl-lacosamide, which is inactive. The metabolite has a longer tmax (0.5-12 hours) and elimination half-life (15-23 hours) than the parent drug. The relative contribution of other CYP isoforms or non-CYP enzymes in the metabolism of lacosamide is unclear. Lacosamide is eliminated primarily by renal excretion and biotransformation.
Adverse Reactions, Warnings, and Contraindications3,4: The most common adverse events reported in trials of lacosamide were diplopia, headache, dizziness, and nausea. As with all other AEDs, lacosamide has the potential to increase the risk of suicidal ideation. Lacosamide has been associated with dose-dependent PR-interval prolongation; thus, caution is advised in patients with known cardiac conduction problems or cardiac disease and in those taking other drugs known to induce PR-interval prolongation. In patients with seizure disorders, lacosamide should be gradually withdrawn (over ≥1 week) to minimize the potential for increased seizure frequency. Rarely, multiorgan hypersensitivity reactions have been reported. If this reaction is suspected, lacosamide should be discontinued.
Drug Interactions3,4: Drug-drug interaction studies have detected no pharmacokinetic interactions between lacosamide and carbamazepine, valproate, digoxin, metformin, omeprazole, or oral contraceptives containing EE and levonorgestrel. There is no evidence of significant interaction between lacosamide and common AEDs. However, the lack of pharmacokinetic interaction does not rule out the possibility of pharmacodynamic interactions, particularly in drugs that affect the cardiac conduction system.
Dosage and Administration3,4: Lacosamide is supplied as 50-mg, 100-mg, 150-mg, and 200-mg tablets and as a 200-mg/20-mL injection. Oral dosing should start at 50 mg twice daily and may be increased to 200 mg to 400 mg/day (recommended maintenance dosing) administered in two divided doses. When patients are switched from oral dosing to IV dosing, the initial total daily IV dose should be equivalent to the total daily oral dose and dosing frequency; IV doses should be infused over a period of 30 to 60 minutes. Patients with mild-to-moderate hepatic impairment or severe renal impairment should receive a maximum of 300 mg/day. Lacosamide use is not recommended in severe hepatic impairment.
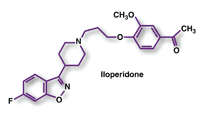
Iloperidone (Fanapt, Vanda Pharmaceuticals)
Indication and Clinical Profile5: Iloperidone has been approved for the treatment of adults with schizophrenia. Its efficacy was demonstrated in two placebo-controlled, short-term trials that used the Positive and Negative Syndrome Scale (PANSS) and the Brief Psychiatric Rating Scale (BPRS) to assess the effects of drug treatment. The first study was a 6-week trial evaluating iloperidone 12 mg to 16 mg/day and iloperidone 20 mg to 24 mg/day (titrated over 7 days) versus placebo and an active control. At the end of this study, both dose ranges of iloperidone were superior to placebo on the endpoint of BPRS total score. In the second trial, iloperidone 24 mg/day was evaluated versus placebo and an active control over 4 weeks. At 4 weeks, iloperidone was superior to placebo on the endpoint of PANSS total score; the efficacy of iloperidone was similar to that of the active control drug.
Pharmacology and Pharmacokinetics5: Similar to other members of the so-called atypical antipsychotic class, iloperidone is proposed to produce its therapeutic effects via a combination of dopamine type 2 (D2) and serotonin type 2 (5-HT2) receptor antagonism. Iloperidone exhibits high-affinity binding to serotonin 5-HT2A and dopamine D2 and D3 receptors (respective Ki values 5.6 nM, 6.3 nM, and 7.1 nM), with relatively low to no affinity for other dopamine and serotonin receptor subtypes or adrenergic and muscarinic receptors.
Iloperidone has a relatively high oral bioavailability (96%) that is not affected by food. Iloperidone is metabolized primarily by carbonyl reduction, hydroxylation (CYP2D6), and O-demethylation (CYP3A4). Approximately 7% to 10% of Caucasian individuals and 3% to 8% of African American individuals lack the capacity to metabolize CYP2D6 substrates and are classified as poor metabolizers (PMs), while others are extensive metabolizers (EMs) or ultrametabolizers. CYP2D6 PMs have higher exposure to iloperidone compared with EMs; thus, dosing adjustments should be considered in this group of patients. Laboratory tests are available to identify CYP2D6 PMs. Similarly, coadministration of iloperidone with strong inhibitors of CYP2D6, such as fluoxetine, results in a 2.3-fold increase in iloperidone plasma exposure, and dosing adjustment is needed. Iloperidone and its metabolites are eliminated primarily in the urine.
Adverse Reactions5: The most common adverse reactions reported for iloperidone in clinical trials were dizziness, dry mouth, fatigue, nasal congestion, orthostatic hypotension (OH), drowsiness, tachycardia, and weight gain. Iloperidone should be used with caution in patients with a history of seizures or a condition that may lower the seizure threshold. Iloperidone has been associated with QTc prolongation. Atypical antipsychotics have been associated with hyperglycemia, some cases of which have been associated with ketoacidosis, hyperosmolar coma, or death. Neuroleptic malignant syndrome, tardive dyskinesias, leukopenia/neutropenia, agranulocytosis, esophageal dysmotility, and aspiration have been reported with antipsychotic drugs, and these reactions may occur with iloperidone. Patients at high risk for suicide should be monitored during iloperidone therapy. Like all atypical antipsychotics, iloperidone has a boxed warning alerting prescribers to an increased risk of death associated with off-label use for the treatment of behavioral problems in elderly patients with dementia-related psychosis.
Drug Interactions5: Both CYP3A4 and CYP2D6 are responsible for iloperidone metabolism; therefore, inhibitors of CYP3A4 (e.g., ketoconazole) or CYP2D6 (e.g., fluoxetine, paroxetine) can inhibit iloperidone clearance, resulting in increased blood levels. Iloperidone should be used with caution with other centrally acting drugs and alcohol. Owing to its alpha1-adrenergic receptor antagonism, iloperidone has the potential to enhance the effect of certain antihypertensive agents. Iloperidone should not be used with other drugs that prolong the QT interval.
Dosage and Administration5: Iloperidone is available as 1-mg, 2-mg, 4-mg, 6-mg, 8-mg, 10-mg, and 12-mg tablets. The drug may be administered without regard to meals. Owing to its alpha-adrenergic blocking properties, iloperidone must be titrated slowly to avoid orthostatic hypotension. The recommended starting dose is 1 mg twice daily. Increases to reach the target effective dosing range of 6 mg to 12 mg twice daily may be made with daily adjustments to 2 mg twice daily, 4 mg twice daily, 6 mg twice daily, 8 mg twice daily, 10 mg twice daily, and 12 mg twice daily on days 2, 3, 4, 5, 6, and 7, respectively. The maximum recommended dose is 12 mg twice daily. Since dose titration is required, full efficacy may not be seen during the first 1 to 2 weeks of treatment. The iloperidone dose should be reduced by one-half when it is administered concomitantly with a strong CYP2D6 inhibitor (fluoxetine, paroxetine) or CYP3A4 inhibitor (ketoconazole, clarithromycin). Iloperidone is not recommended in patients with hepatic impairment.
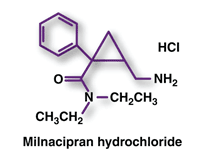
Milnacipran (Savella, Forest Pharmaceuticals)
Indication and Clinical Profile6,7: Milnacipran, a selective serotonin norepinephrine reuptake inhibitor (SNRI), has been approved by the FDA for the management of fibromyalgia. Fibromyalgia, which is estimated to affect more than six million Americans, is a chronic, debilitating condition characterized by widespread pain and decreased physical functioning. The efficacy of milnacipran for the treatment of fibromyalgia was established in two phase III clinical trials involving 2,084 patients (milnacipran, n = 1,460; placebo, n = 624). Both studies demonstrated clinically significant improvements in fibromyalgia symptoms compared with placebo.
Pharmacology and Pharmacokinetics6,7: Milnacipran blocks the reuptake of norepinephrine and serotonin, with greater selectivity for the inhibition of norepinephrine reuptake in vitro. These two neurotransmitters are thought to a play a central role in the symptoms of fibromyalgia. Milnacipran is well absorbed (bioavailability >85%), producing a Cmax at 2 to 4 hours and steady-state levels at 36 to 48 hours. It undergoes minimal metabolism by glucuronidation and desethylation. The active enantiomer, d-milnacipran, has a longer elimination half-life (8-10 hours) than the l-enantiomer (4-6 hours). The drug is eliminated primarily in the urine (55%), necessitating dose adjustment in severe renal impairment.
Adverse Reactions, Warnings, and Precautions6,7: The most common adverse events associated with milnacipran are nausea, constipation, hot flush, hyperhidrosis, vomiting, palpitations, increased heart rate, dry mouth, and hypertension.
Milnacipran is an SNRI and is similar to agents used to treat depression and other psychiatric disorders. Therefore, it carries the same boxed warning concerning increased risk of suicidal thinking/behavior in children, adolescents, and young adults. Milnacipran should be used with caution in patients with a history of seizure disorder, mania, or controlled narrow-angle glaucoma (NAG). The use of milnacipran is contraindicated in patients with uncontrolled NAG. Milnacipran can affect urethral resistance and micturition and cause mydriasis. Patients with substantial alcohol use or evidence of chronic liver disease should not receive the drug.
Milnacipran treatment has been associated with increased liver enzymes, fulminant hepatitis (rarely), and hyponatremia. These conditions may require drug discontinuation. Milnacipran is pregnancy category C, and use during labor and delivery is not recommended. Milnacipran contains FD&C Yellow No. 5, which may cause allergic-type reactions in susceptible individuals.
Drug Interactions6,7: Milnacipran is contraindicated in patients who are taking monoamine oxidase inhibitors (MAOIs) or are within 14 days of discontinuing an MAOI. Coadministration of milnacipran with serotonergic drugs or serotonin precursors is not recommended owing to the potential for development of serotonin syndrome. Milnacipran may increase the risk of bleeding; concurrent use with drugs that affect coagulation may add to this risk. The antihypertensive effect of clonidine may be inhibited by milnacipran. Clinically significant cytochrome-related drug interactions are unlikely.
Dosage and Administration6,7: Milnacipran is available as 12.5-mg, 25-mg, 50-mg, and 100-mg tablets. The usual dose is 100 mg/day (50 mg twice daily). Dosing should be titrated according to the following schedule: day 1, 12.5 mg once; days 2-3, 25 mg/day (12.5 mg twice daily); days 4-7, 50 mg/day (25 mg twice daily); after day 7, 100 mg/day (50 mg twice daily). Based on individual patient response, the dose may be increased to 200 mg/day (100 mg twice daily). Maintenance doses in severe renal impairment should be reduced to 50 mg/day (25 mg twice daily). Milnacipran should be tapered and not abruptly discontinued after extended use.
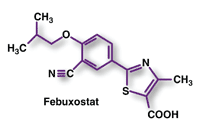
Febuxostat (Uloric, Takeda)
Indication and Clinical Profile8,9: Febuxostat has been approved for the chronic management of hyperuricemia in patients with gout. This once-daily oral medication is the first new treatment option for gout in more than 40 years. Gout is the most common type of inflammatory arthritis in men over the age of 40, affecting 5.1 million Americans. It is a chronic, often progressive condition characterized by attacks, or "flares," that are marked by intense pain, heat, redness, and swelling in the affected joint. These symptoms result from an acute inflammatory response to elevated uric acid levels in the joints. Thus, the goal in treating chronic gout is the reduction of serum uric acid levels and maintenance at <6 mg/dL.
The efficacy of febuxostat was examined in three randomized, double-blind, controlled trials involving 3,402 subjects. In all three studies, febuxostat 80 mg/day was superior to allopurinol in the proportion of patients who achieved serum uric acid <6 mg/dL at the final visit. Febuxostat 40 mg/day was not superior to allopurinol, but it was as effective in lowering serum uric acid to <6 mg/dL.
Pharmacology and Pharmacokinetics8,9: Like allopurinol, febuxostat lowers plasma uric acid levels by inhibiting the enzyme xanthine oxidase (XO). XO is responsible for the breakdown of the purine metabolite hypoxanthine to xanthine and then to uric acid. By blocking XO, febuxostat slows the rate of uric acid production, thereby lowering elevated levels of serum uric acid.
Febuxostat is about 50% absorbed, giving a Cmax at 1 to 1.5 hours. It is metabolized by conjugation via the uridine diphosphate glucuronosyltransferase (UGT) isoenzymes UGT1A1, 1A3, 1A9, and 2B7 and by oxidation via CYP450 isozymes 1A2, 2C8, and 2C9. Metabolism leads to four active metabolites. Febuxostat undergoes hepatic (45%) elimination and renal (49%) elimination.
Adverse Reactions, Warnings, and Contraindications8,9: The most commonly reported adverse reactions for febuxostat in clinical trials were liver-function abnormalities (>3 times upper limit of normal), nausea, joint pain, rash, and gout flares. Periodic monitoring of liver function is recommended. A higher rate of cardiovascular thromboembolic events was observed in patients treated with febuxostat (0.74 per 100 patient-years) compared with allopurinol (0.60 per 100 patient-years) in clinical trials, but a causal relationship has not been established. Thus, monitoring for signs and symptoms of myocardial infarction and stroke is advised. Febuxostat is pregnancy category C. The agent is contraindicated in patients receiving azathioprine, mercaptopurine, or theophylline (see Drug Interactions).
Drug Interactions8,9: Drug-interaction studies involving febuxostat and other drugs that are metabolized by XO have not been conducted. However, inhibition of XO by febuxostat may cause increased plasma concentrations of these drugs, thus leading to toxicity. Febuxostat therefore is contraindicated in patients who are receiving azathioprine, mercaptopurine, or theophylline. Based on drug-interaction studies, febuxostat does not have clinically significant interactions with colchicine, naproxen, indomethacin, hydrochlorothiazide, warfarin, or desipramine. Drug-interaction studies of febuxostat with cytotoxic chemotherapy have not been conducted.
Dosage and Administration8,9: Febuxostat is supplied as 40-mg and 80-mg tablets. The drug may be taken without regard to food or antacid use. The recommended starting dose is 40 mg once daily. The dose may be increased to 80 mg/day in patients who do not achieve a serum uric acid level <6 mg/dL after 2 weeks on the lower dose. No dose adjustments are required in patients with mild-to-moderate renal or hepatic impairment. Caution is recommended if febuxostat is administered in severe renal or hepatic impairment.
REFERENCES
1. Banzel (rufinamide) product information. Woodcliff Lake, NJ: Eisai Inc; November 2008.
2. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950.
3. Vimpat (lacosamide) product information. Smyrna, GA: UCB, Inc; January 2009.
4. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
5. Fanapt (iloperidone) product information. Rockville, MD: Vanda Pharmaceuticals, Inc; May 2009.
6. Savella (milnacipran HCl) product information. St. Louis, MO: Forest Pharmaceuticals, Inc; March 2009.
7. Mease PJ, Clauw DJ, Gendreau RM, et al. The efficacy and safety of milnacipran for treatment of fibromyalgia. a randomized, double-blind, placebo-controlled trial. J Rheumatol. 2009;36:398-409.
8. Uloric (febuxostat) product information. Deerfield, IL: Takeda Pharmaceuticals America, Inc; February 2009.
9. Schumacher HR Jr, Becker MA, Lloyd E, et al. Febuxostat in the treatment of gout: 5-yr findings of the FOCUS efficacy and safety study. Rheumatology (Oxford). 2009;48:188-194.



