US Pharm. 2014;39(10)20-26.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2013–2014 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Umeclidinium and Vilanterol (Anoro Ellipta, GlaxoSmithKline)
Indication and Clinical Profile1,2: The combination of umeclidinium (anticholinergic) and vilanterol (long-acting beta2-adrenergic agonist [LABA]) is approved for long-term maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD). COPD is a lung disease involving chronic bronchitis and/or emphysema that is characterized by airflow obstruction that interferes with breathing and worsens over time. It is typically caused by long-term exposure to lung irritants that damage the lungs and airways. Patients develop COPD at age ≥40 years and typically experience symptoms of chest tightness, chronic cough, and excessive phlegm. According to the National Heart, Lung, and Blood Institute, COPD is currently the third leading cause of death in the United States. Umeclidinium/vilanterol inhalation powder is the first combination product comprising two long-acting bronchodilators for once-daily use to receive approval in the United States for maintenance treatment of COPD.
Approval of umeclidinium/vilanterol was based primarily on six dose-ranging trials and two placebo-controlled confirmatory trials. The dose-ranging trials, which included 1,908 subjects with COPD or asthma, were conducted for the purpose of determining dosage selection for the later confirmatory trials. From these trials, dosages of once-daily umeclidinium/vilanterol 62.5 mcg/25 mcg and 125 mcg/25 mcg were selected to be evaluated in the confirmatory COPD trials, which enrolled 4,733 subjects aged ≥40 years with a history of smoking and were conducted over a 6-month period. The first trial compared umeclidinium/vilanterol 62.5 mcg/25 mcg, umeclidinium 62.5 mcg, vilanterol 25 mcg, and placebo. The primary endpoint was the change from baseline predose forced expiratory volume in 1 second (FEV1) at day 169, 23 to 24 hours after the dose from the previous day, compared with placebo or with umeclidinium 62.5 mcg or vilanterol 25 mcg alone. Umeclidinium/vilanterol 62.5 mcg/25 mcg met this endpoint by demonstrating a greater increase in mean change from baseline in trough (predose) FEV1 relative to either drug alone or placebo. The second trial was of the same design; it compared umeclidinium/vilanterol 125 mcg/25 mcg, umeclidinium 125 mcg, vilanterol 25 mcg, and placebo and demonstrated results analogous to those of the first trial. Umeclidinium/vilanterol approval was also supported by two active-control trials and two crossover trials involving 5,388 subjects with COPD.
Pharmacology and Pharmacokinetics1,2: Umeclidinium (FIGURE 1) is a long-acting competitive antagonist at muscarinic receptors M1, M3, and M5. By blocking M3 receptors on smooth muscle in the airways, umeclidinium blocks bronchoconstriction via cholinergic pathways, resulting in net bronchodilation. Vilanterol (FIGURE 1) is an LABA that stimulates adenylate cyclase, catalyzing the conversion of adenosine triphosphate to cyclic 3´,5´-adenosine monophosphate (cAMP). Generation of cAMP in relaxation of bronchial smooth muscle results in relaxation and inhibition of release of immediate hypersensitivity mediators from cells such as mast cells.
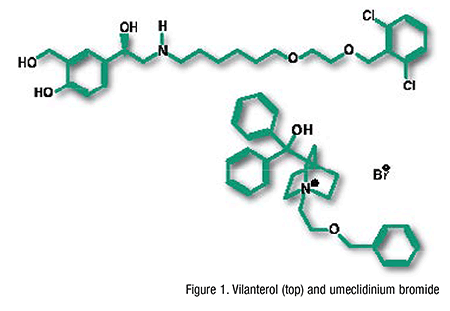
Umeclidinium provides peak plasma concentrations at 5 to 15 minutes; it has a half-life of 11 hours and is 89% plasma protein–bound. The drug is primarily metabolized to various hydroxylation or O-dealkylation products, with varying activities via CYP2D6 followed by glucuronidation. Umeclidinium does not appear to significantly induce or inhibit metabolism by any major cytochrome isozyme. It is eliminated both renally (22%) and fecally (58%).
Vilanterol provides peak plasma concentrations at 5 to 15 minutes; it has a half-life of 11 hours and is 94% plasma protein–bound. The drug is metabolized to a range of metabolites, with significantly reduced beta1- and beta2-agonist activity via CYP3A4. Vilanterol does not appear to significantly induce or inhibit metabolism by any major cytochrome isozyme. It is eliminated primarily via renal excretion (70%), along with some fecal elimination (30%).
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions reported in clinical trials included pharyngitis, sinusitis, lower respiratory tract infection, constipation, diarrhea, pain in extremities, muscle spasms, neck pain, and chest pain. Other more serious effects, including worsening of urinary retention, paradoxical bronchospasm, acute narrow-angle glaucoma, and cardiovascular effects, also were noted during trials. The drug’s label carries a boxed warning stating that umeclidinium/vilanterol is not approved for the treatment of asthma, as LABAs have been shown to increase the risk of asthma-related death. The warning also states that the drug should not be used as a rescue therapy to treat acute bronchospasm. Umeclidinium/vilanterol has not been studied in pregnant women and should be avoided during pregnancy unless the potential benefit justifies the potential risk to the fetus (Pregnancy Category C).
Use of beta-blockers should be avoided in patients taking umeclidinium/vilanterol, as the beta-blocker may antagonize the bronchodilatory effects of vilanterol. Also, coadministration of monoamine oxidase inhibitors and tricyclic antidepressants with strong inhibitors of CYP3A4 (e.g., ketoconazole) may potentiate the effect of vilanterol on the cardiovascular system; therefore, these agents should be used together only with extreme caution. The electrocardiographic changes and hypokalemia often associated with loop and thiazide diuretics may worsen if the diuretic is taken concomitantly with vilanterol. Also, the use of other anticholinergics should be avoided, as this may potentiate the actions of umeclidinium. No clinically significant difference in systemic exposure to umeclidinium was reported in the presence of CYP2D6 inhibitors.
Dosage and Administration1,2: Umeclidinium/vilanterol is supplied as a powder formulation for oral inhalation containing 62.5 mcg of umeclidinium and 25 mcg of vilanterol. The recommended dosage is one inhalation daily. No dosage adjustment is recommended for elderly patients or those with renal or mild-to-moderate hepatic impairment. The safety and efficacy of umeclidinium/vilanterol has not been established in pediatric patients or in patients with severe hepatic impairment. Umeclidinium/vilanterol comes with a patient medication guide that includes information regarding potential risks of taking the drug and instructions for proper use.
Vortioxetine (Brintellix, Takeda Pharmaceuticals America, Lundbeck)
Indication and Clinical Profile3,4: Vortioxetine is a new antidepressant approved to treat adults with major depressive disorder (MDD). MDD is predominantly characterized by a loss of interest in usual activities and mood changes that typically interfere with the person’s professional, personal, and social life by causing significant changes in appetite, sleep habits, and cognition. MDD also may result in increased feelings of guilt or worthlessness and suicidal thoughts. Some patients with MDD experience only a single episode of depression; however, frequently patients experience multiple recurrences throughout their lifetime.
FDA approval of vortioxetine was based on six 6- to 8-week randomized, double-blind, placebo-controlled, fixed-dose studies involving 2,390 adult patients with MDD. Vortioxetine’s efficacy versus placebo was based on patients’ mean change from baseline to endpoint in Hamilton Depression Scale score in study 2, and on the Montgomery-Asberg Depression Rating Scale in all other studies. In all but two studies, patients receiving vortioxetine experienced a significant improvement in depressive symptoms compared with placebo. In the two studies that failed to show effectiveness, the dosage used was 5 mg. In a 24- to 64-week maintenance study in 639 adults with MDD, patients receiving vortioxetine experienced a statistically significant decrease in frequency of recurrent depressive episodes.
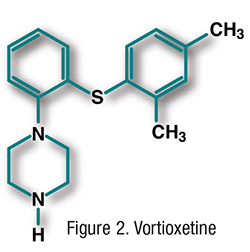
Pharmacology and Pharmacokinetics3,4: Vortioxetine (FIGURE 2) is a serotonin modulator and stimulator that is thought to exhibit its antidepressant effect by functioning as an inhibitor of serotonin (5-HT) reuptake pumps. It also acts as an antagonist at 5-HT3 receptors and an agonist at 5-HT1A receptors. The absolute bioavailability of vortioxetine is 75% when the drug is taken with or without food. Vortioxetine produces peak plasma concentrations at 7 to 11 hours and has a half-life of 66 hours. It undergoes oxidative metabolism via CYP2D6, -3A4/5, -2C19, -2C9, -2A6, -2C8, and -2B6, with its major inactive metabolite being formed via CYP2D6. Vortioxetine does not appear to significantly induce or inhibit metabolism by any major cytochrome isozyme. About 59% of the dose is excreted in the urine and 26% in the feces as metabolites.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions reported in clinical trials were nausea, constipation, vomiting, xerostomia, dizziness, and sexual dysfunction. The drug’s label has a boxed warning concerning the risk of suicidal thoughts and behavior in children, adolescents, and young adults aged 18 to 24 years during initial treatment. Patients and caregivers should alert a healthcare professional immediately if changes in mood or behavior that are atypical for the patient, such as agitation, irritability, or suicidal thoughts, are observed. Vertigo, dyspepsia, dysgeusia, flushing, and activation of mania were also seen in a few patients taking vortioxetine. Patients should be cautioned about the risk of increased incidence of bleeding and hyponatremia with serotonergic drugs.
Vortioxetine is metabolized by CYP2D6; therefore, concurrent use of potent CYP2D6 inhibitors (e.g., bupropion, fluoxetine, paroxetine, quinidine) can increase serum concentrations. The vortioxetine dosage should be reduced by half if these drugs are taken concurrently. Potent inducers of CYP2D6 (e.g., carbamazepine, rifampicin, phenytoin) decrease vortioxetine levels and require an increase in the vortioxetine dosage. Patients taking vortioxetine while on other drugs that affect serotonergic neurotransmission (e.g., selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, triptans, buspirone) are at increased risk for serotonin syndrome and should be monitored for symptoms. Concomitant use of vortioxetine and drugs that interfere with hemostasis (e.g., nonsteroidal anti-inflammatory drugs, aspirin, warfarin) may result in prolonged bleeding time. Vortioxetine is contraindicated in patients currently taking monoamine oxidase inhibitors (MAOIs). Vortioxetine therapy should not be initiated within 14 days after discontinuing an MAOI, and it is recommended that a patient not be put on an MAOI until 21 days after discontinuation of vortioxetine. Vortioxetine has demonstrated feticidal and teratogenic effects in animals and should not be used during pregnancy unless the potential benefit to the patient offsets the potential risk to the fetus (Pregnancy Category C).
Dosage and Administration3,4: Vortioxetine is supplied as 5-mg, 10-mg, 15-mg, and 20-mg tablets for oral administration, and the recommended starting dosage is 10 mg once daily. The dosage should then be increased to 20 mg as tolerated, or to 10 mg per day in poor metabolizers of CYP2D6. Patients who cannot tolerate higher dosages of vortioxetine may take 5 mg per day. No dosage adjustments are recommended in patients with renal impairment or mild-to-moderate hepatic impairment or in elderly patients. It is recommended that, in patients taking ≥15 mg daily, the dosage should be reduced to 10 mg daily for 1 week before vortioxetine is discontinued.
Dapagliflozin (Farxiga, Bristol-Myers Squibb, AstraZeneca)
Indication and Clinical Profile5,6: Dapagliflozin is a new sodium-glucose cotransporter 2 (SGLT2) inhibitor approved as an adjunct to diet and exercise to improve glycemic control in patients with type 2 diabetes mellitus (DM2). DM2 is a chronic, progressive disease affecting more than 23 million people in the United States. Even with diet and exercise, patients often must take multiple medications to help manage their blood-glucose levels. Dapagliflozin is the second drug of its type to be approved under this indication, the first being canagliflozin (Invokana).
FDA approval of dapagliflozin was based on two pools of 12 and 13 placebo-controlled studies involving 2,338 and 2,360 patients, respectively. Throughout these studies, dapagliflozin resulted in significant improvements in mean change from baseline in A1C versus placebo both as monotherapy and as add-on therapy to metformin, glimepiride, pioglitazone, sitagliptin, or insulin. Other studies found that dapagliflozin was noninferior to glipizide as an add-on to metformin, and that initial treatment with dapagliflozin and metformin was more effective than metformin alone in improving baseline A1C.
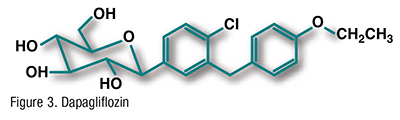
Pharmacology and Pharmacokinetics5,6: SGLT2 (FIGURE 3) is a renal transport protein that is responsible for the majority of glucose reabsorption from the proximal renal tubule. Inhibition of SGLT2 by dapagliflozin results in increased glucose excretion and an overall reduction in blood-glucose levels. Dapagliflozin 10 mg has an oral bioavailability of 78% and a terminal half-life of approximately 12.9 hours. The drug is 91% plasma protein–bound. Its primary route of metabolism is by glucuronidation. Dapagliflozin does not induce or inhibit CYP450 isoenzymes. About 75% of the dose is excreted in the urine and 21% in the feces; <2% is excreted unchanged in the urine.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions (≥5%) reported in clinical trials were female genital fungal infections, urinary tract infections, and nasopharyngitis. Dapagliflozin produced hypoglycemia at a rate comparable to that for placebo. Because of its diuretic effect, adverse reactions such as dehydration, hypovolemia, and hypotension can occur, particularly in elderly patients with renal dysfunction and in those taking loop diuretics. Dapagliflozin may increase serum creatinine and decrease epidermal growth factor receptor (EGFR) and cause an increase in LDL cholesterol (LDL-C). Therefore, LDL-C should be monitored after treatment initiation in patients taking dapagliflozin. In clinical trials, patients receiving dapagliflozin had a higher incidence of bladder cancer than those who received placebo. Because of this, patients with active bladder cancer should not use dapagliflozin, and those with a history of prior bladder cancer should consider potential benefits and risks before use. Dapagliflozin may affect renal development in the fetus primarily during the late second trimester and third trimester; therefore, dapagliflozin should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus after appropriate alternative therapies have been considered (Pregnancy Category C).
The FDA is requiring postmarketing studies for dapagliflozin to evaluate cardiovascular (CV) risk in patients with high baseline CV disease risk; the risk of bladder cancer; and the drug’s pharmacokinetics, safety, and efficacy in pediatric patients. It is also mandating a program to monitor liver abnormalities and pregnancy outcomes in treated patients.
Dapagliflozin has no significant interactions with drugs that are CYP450 substrates or inhibitors, or with drugs excreted renally. Patients taking insulin or an insulin secretagogue in combination with dapagliflozin may have to lower the insulin or insulin secretagogue dosage to reduce the risk of hypoglycemia.
Dosage and Administration5,6: Dapagliflozin is supplied as 5-mg and 10-mg tablets. The recommended dosage is 5 mg every morning, with or without food. In patients who require additional glycemic control, the dosage may be increased to 10 mg once daily. Dapagliflozin should not be used in patients with moderate-to-severe renal impairment, and renal function should be assessed prior to initiation and during therapy. Dapagliflozin should be discontinued if EGFR is persistently <60 mL/min/1.73 m2 during treatment. No dosage adjustment is recommended in elderly patients or in patients with mild renal impairment or mild-to-moderate hepatic impairment. Dapagliflozin has not been studied in pediatric patients or in patients with severe hepatic impairment.
Luliconazole (Luzu, Medicis)
Indication and Clinical Profile7,8: The FDA approved luliconazole 1% cream for the topical treatment of athlete’s foot (interdigital tinea pedis), jock itch (tinea cruris), and ringworm (tinea corporis) caused by the fungal pathogens Trichophyton rubrum and Epidermophyton floccosum in patients aged ≥18 years. Luliconazole is the first product approved for 1-week, once-daily treatment of common skin diseases caused predominantly by dermatophytic fungi. Other options require 2 weeks of therapy.
Luliconazole has been extensively studied, with three pivotal studies forming the basis for FDA approval. Two of these studies (study 1 and study 2) enrolled 423 patients with tinea pedis who were randomized to luliconazole or a placebo vehicle. Luliconazole or vehicle was applied to the entire area of the forefoot, including all interdigital web spaces and approximately 2.5 cm (1 inch) of the surrounding area of the foot once daily for 14 days. Overall treatment success was defined as complete clearance (clinical cure and mycological cure) at 4 weeks post treatment. In study 1, 26% of subjects treated with luliconazole were completely cleared of infection, compared with 2% of those treated with vehicle. In study 2, 14% of subjects treated with luliconazole were completely cleared, compared with 3% of those treated with vehicle. The pivotal study for tinea cruris included 256 patients who were randomized to receive luliconazole or vehicle cream to the affected area once daily for 7 days. Overall treatment success was defined as complete clearance (clinical and mycological cure) at 3 weeks post treatment. After 1 week of treatment, 21% of subjects treated with luliconazole were completely cleared, compared with 4% of those treated with vehicle.
Pharmacology and Pharmacokinetics7,8: Luliconazole (FIGURE 4) is an azole antifungal that blocks fungal ergosterol biosynthesis by inhibiting the enzyme lanosterol demethylase. Inhibition of this enzyme’s activity by azole drugs results in decreased amounts of ergosterol—a constituent of fungal cell membranes—and a corresponding accumulation of lanosterol.
Although luliconazole is applied topically, pharmacokinetic studies have demonstrated that there is some systemic absorption. In clinical trials, plasma concentrations of luliconazole on day 15 were measurable in all tinea pedis and tinea cruris subjects and fluctuated little during the 24-hour interval. Plasma levels range from approximately 1 to 7 ng/mL, with exposures as expressed by AUC0-24 of 18 to 120 ng · h/mL.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions were mild application-site reactions, reported in <1% of subjects for both luliconazole and vehicle. There have been some reports of contact dermatitis and cellulitis during postmarketing use, but since these reactions are reported voluntarily by a population of uncertain size, it is not always possible to reliably estimate frequency or establish a causal relationship to drug exposure. Luliconazole is a Pregnancy Category C drug, and it is not known whether luliconazole is excreted in human milk. Therefore, caution should be exercised in pregnant and nursing women, and ideally the drug should be used only if the potential benefit justifies the potential risk to the fetus or child.
Because of the drug-interaction potential of azole antifungals, luliconazole was assessed in vitro for its ability to inhibit a number of cytochrome enzymes. In these studies, luliconazole at therapeutic doses—particularly when used in patients with moderate-to-severe tinea cruris—showed some inhibitory activity toward CYP2C19 and -3A4. However, no in vivo drug-interaction trials have been conducted to evaluate the effect of luliconazole on other drugs that are substrates of these CYP isozymes. Luliconazole is not expected to inhibit CYP1A2, -2C9, or -2D6, based on in vitro assessment. The induction potential of luliconazole on CYP enzymes has not been evaluated.
Dosage and Administration7,8: Luliconazole is supplied as a 1% cream for topical administration and contains 10 mg of the active drug per gram. For interdigital tinea pedis, a thin layer should be applied to the affected and immediate surrounding areas (1 in.) once daily for 2 weeks. For tinea cruris and tinea corporis, the cream should be applied to the affected skin and immediate surrounding areas once daily for 1 week. The drug is intended for topical use only, and not for ophthalmic, oral, or intravaginal application.
Sofosbuvir (Sovaldi, Gilead Sciences)
Indication and Clinical Profile9,10: The FDA approved sofosbuvir to treat chronic hepatitis C virus (HCV) infection. Sofosbuvir is the first drug that has demonstrated safety and efficacy to treat certain types of HCV infection without the need for coadministration of interferon. This approval represents a significant shift in the treatment paradigm for some patients with chronic hepatitis C. Sofosbuvir is the third drug with breakthrough therapy designation to receive FDA approval; the first two are the chronic lymphocytic leukemia drugs obinutuzumab (Gazyva) and ibrutinib (Imbruvica). Simeprevir (Olysio) was approved to treat chronic HCV infection in November 2013, but not as a breakthrough therapy.
Hepatitis C is a viral disease that causes inflammation of the liver, which can lead to diminished liver function or liver failure. Most people infected with HCV have no symptoms until liver damage is apparent, which may take several years. Some people with chronic HCV infection develop scarring and poor liver function (cirrhosis) over many years, which can result in complications such as bleeding, jaundice, ascites, infections, or liver cancer. According to the CDC, about 3.2 million Americans are infected with HCV. There are several different genotypes of HCV infection, and the prevalence of HCV genotypes in the United States is 79% genotype 1, 13% genotype 2, 6% genotype 3, and 2% genotypes 4, 5, and 6 combined. Depending on the type of HCV infection, the treatment regimen could include sofosbuvir and ribavirin or sofosbuvir, ribavirin, and peginterferon alfa. Ribavirin and peginterferon alfa are also used to treat HCV infection.
The effectiveness of sofosbuvir was demonstrated in six clinical trials involving 1,947 participants not previously treated for their disease (treatment-naïve) or not responsive to previous treatment (treatment-experienced), including patients coinfected with HCV and HIV. The trials were designed to measure whether HCV was no longer detected in the blood ≥12 weeks after treatment cessation (sustained virologic response), which would suggest that a participant’s HCV infection has been cured. Results of all clinical trials showed that a treatment regimen containing sofosbuvir was effective in treating multiple types of HCV. Additionally, sofosbuvir demonstrated efficacy in participants who could not tolerate or take an interferon-based treatment regimen and in participants with liver cancer awaiting liver transplantation, addressing unmet medical needs in these populations.
Pharmacology and Pharmacokinetics9,10: Sofosbuvir (FIGURE 5) is an inhibitor of the HCV nonstructural protein 5B (NS5B) RNA-dependent RNA polymerase, which is essential for viral replication. It is a nucleotide prodrug that undergoes intracellular metabolism to form the pharmacologically active uridine analogue triphosphate, which can be incorporated into HCV RNA by the NS5B polymerase and acts as a chain terminator, similar to antiretroviral drugs. Sofosbuvir has potent activity against all HCV genotypes (1-6), including strains resistant to protease inhibitors.
Following oral administration, sofosbuvir is rapidly absorbed, providing peak plasma concentrations at 0.5 to 2 hours post dose, irrespective of dose. Peak plasma concentration of sofosbuvir’s primary metabolite, GS-331007, occurs 2 to 4 hours post dose. Absorption does not appear to be altered by coadministration with food. Sofosbuvir undergoes extensive hepatic metabolism to form the pharmacologically active nucleoside analogue triphosphate (GS-461203). The metabolic-activation pathway involves sequential hydrolysis of the carboxyl ester moiety catalyzed by human cathepsin A or carboxylesterase 1 and phosphoramidate cleavage by histidine triad nucleotide-binding protein 1, followed by phosphorylation by the pyrimidine nucleotide biosynthesis pathway. Dephosphorylation results in the formation of nucleoside metabolite GS-331007, which cannot be efficiently rephosphorylated and lacks anti-HCV activity in vitro. Sofosbuvir is eliminated primarily in the urine (80%), mainly as its GS-331007 metabolite. The median terminal half-lives of sofosbuvir and GS-331007 are reported to be 0.4 and 27 hours, respectively. No dosage adjustment is required for patients with mild-to-moderate renal or hepatic impairment.
Adverse Reactions and Drug Interactions9,10: The most common adverse reactions in clinical-study participants treated with sofosbuvir and ribavirin were fatigue and headache. In participants treated with sofosbuvir, ribavirin, and peginterferon alfa, the most common adverse reactions were fatigue, headache, nausea, insomnia, and anemia. Sofosbuvir is not teratogenic in animals, but it has not been studied in pregnant women. However, ribavirin is teratogenic and embryotoxic, and peginterferon has abortifacient effects (Pregnancy Category X). Therefore, pregnancy in female patients and female partners of male patients should be avoided during treatment with this regimen. Patients must have a negative pregnancy test prior to therapy initiation, use at least two effective nonhormonal methods of contraception, and have monthly pregnancy tests.
Sofosbuvir is a substrate of P-glycoprotein (Pgp) and should not be administered with potent Pgp inducers such as rifampin. Unlike protease inhibitors, sofosbuvir and its metabolites are not inhibitors, inducers, or substrates of CYP450 enzymes.
Dosage and Administration9,10: Sofosbuvir is supplied as tablets for oral administration and should be used in combination with ribavirin or with pegylated interferon and ribavirin. The recommended dosage of sofosbuvir is 400 mg orally once daily with or without food. For patients with HCV genotype 1 or 4 infection, sofosbuvir should be given with peginterferon and ribavirin for 12 weeks. In patients with HCV genotype 1 infection who cannot take interferon, the use of sofosbuvir plus ribavirin for 24 weeks may be considered as an alternative. Sofosbuvir should be administered with ribavirin for 12 weeks in patients infected with HCV genotype 2, and for 24 weeks in those infected with HCV genotype 3. The cost of a 28-tablet bottle of sofosbuvir is $28,000; accordingly, a 12-week supply of the drug would cost $84,000 and a 24-week supply would cost $168,000.
REFERENCES
1. Anoro Ellipta (umeclidinium and vilanterol inhalation powder) product information. Research Triangle Park, NC: GlaxoSmithKline; May 2014.
2. Donohue JF, Maleki-Yazdi MR, Kilbride S, et al. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107:1538-1546.
3. Brintellix (vortioxetine) product information. Deerfield, IL: Takeda Pharmaceuticals America; July 2014.
4. Jain R, Mahableshwarkar AR, Jacobsen PL, et al. A randomized, double-blind, placebo-controlled 6-wk trial of the efficacy and tolerability of 5 mg vortioxetine in adults with major depressive disorder. Int J Neuropsychopharmacol. 2013;16:313-321.
5. Farxiga (dapagliflozin) product information. Princeton, NJ: Bristol-Myers Squibb Co; January 2014.
6. Bailey CJ, Iqbal N, T’Joen C, List JF. Dapagliflozin monotherapy in drug-naïve patients with diabetes: a randomized-controlled trial of low-dose range. Diabetes Obes Metab. 2012;14:951-959.
7. Luzu (luliconazole) product information. Bridgewater, NJ: Medicis; November 2013.
8. Jarratt M, Jones T, Kempers S, et al. Luliconazole for the treatment of interdigital tinea pedis: a double-blind, vehicle-controlled study. Cutis. 2013;91:203-210.
9. Sovaldi (sofosbuvir) product information. Foster City, CA: Gilead Sciences, Inc; December 2013.
10. Sulkowski MS, Rodriguez-Torres M, Lalezari JP, et al. All-oral therapy with sofosbuvir plus ribavirin for the treatment of HCV genotype 1, 2, and 3 infection in patients co-infected with HIV (PHOTON-1). In: Program and abstracts of the 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1-5, 2013; Washington, DC. Abstract 212.
To comment on this article, contact rdavidson@uspharmacist.com.



