US Pharm. 2015;40(10):39-44.
ABSTRACT: New molecular entities (NMEs), as defined by the FDA, are drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in the past year (TABLE 1) detail the basic clinical and pharmacologic profiles for each new drug, as well as its pharmacokinetics, adverse reactions, drug interactions, and dosing data. Note that the information for each NME was obtained primarily from sources published prior to FDA approval; thus, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature, such as the emergence of additional adverse reactions and boxed warnings.
Secukinumab (Cosentyx, Novartis Pharmaceuticals)
Indication and Clinical Profile1,2: Secukinumab is the first interleukin-17A (IL-17A) inhibitor FDA-approved to treat adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Psoriasis is a chronic inflammatory autoimmune-based skin condition that causes patches of skin redness, irritation, and discomfort. It affects about 7.5 million Americans and occurs more commonly in patients with a family history of the disease, with onset usually between the ages of 15 and 35 years. The most common form of psoriasis is plaque psoriasis, in which patients develop thick, red skin with flaky, silver-white patches called scales.
Topical preparations, including corticosteroids and vitamin D analogues, are first-line treatment for mild disease, and ultraviolet B (UVB) phototherapy is used when the disease is more widespread or unresponsive to topical therapies. For patients with moderate-to-severe disease, systemic treatments are available including methotrexate, cyclosporine, the oral retinoid acitretin, and biologic therapies including tumor necrosis factor (TNF) inhibitors, interleukin antagonists, and now secukinumab.
Secukinumab’s safety and efficacy were demonstrated in four clinical trials with a total of 2,403 plaque psoriasis patients (aged ≥18 years) who were candidates for phototherapy or systemic therapy with a minimum body surface area involvement of 10%, and a Psoriasis Area and Severity Index (PASI) score ≥12. Participants were randomly assigned to receive secukinumab (300 or 150 mg) or placebo and, in one trial, etanercept as a biologic active control. In all trials, the endpoints were the proportion of subjects who achieved a reduction in PASI score of at least 75% (PASI 75) from baseline to week 12 and treatment success (clear or almost clear) on the Investigator’s Global Assessment (IGA). In these studies, secukinumab met all primary and key secondary endpoints, including PASI 75 and 90 and IGA responses, showing significant skin clearance at week 12.
Pharmacology and Pharmacokinetics1,2: Secukinumab is a human immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds to the IL-17A cytokine and inhibits its interaction with the IL-17 receptor. IL-17A is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. By binding to IL-17A, secukinumab prevents it from binding to its receptor and inhibits its ability to trigger the inflammatory response that plays a role in the development of plaque psoriasis, including the release of proinflammatory cytokines and chemokines.
Following multiple SC doses, the mean serum trough concentrations of secukinumab ranged from 22.8 (150-mg dose) to 45. 2 mcg/mL (300-mg dose) at week 12. Steady-state concentrations were achieved by week 24 following the every-4-week dosing regimen. The metabolic pathway of secukinumab has not been characterized, but as a human IgG1-kappa monoclonal antibody it is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG. The mean systemic clearance (CL) ranged from 0.14 to 0.22 L/day and the mean half-life ranged from 22 to 31 days in subjects in plaque psoriasis trials. No formal trials evaluating the effect of hepatic or renal impairment on the pharmacokinetics of secukinumab have been conducted.
Adverse Reactions and Drug Interactions1,2: The most common adverse effects of secukinumab in clinical trials included nasopharyngitis, diarrhea, and upper respiratory infection. Infections occurred at a higher rate in secukinumab-treated patients than in those who received placebo. Thus, caution should be exercised when considering the use of secukinumab in patients with a chronic infection or history of recurrent infection, and patients should be screened for tuberculosis before starting therapy. In addition, exacerbations of Crohn’s disease and hypersensitivity reactions were reported during clinical trials in patients taking secukinumab. Secukinumab is being approved with a Medication Guide to inform patients of these risks. Formal drug interaction studies have not been conducted, but patients treated with secukinumab should not receive live vaccines. No treatment-related effects on functional, morphological, or immunologic development were observed in fetuses in animal models; thus, secukinumab is classified as a Pregnancy Category B drug.
A role for IL-17A in the regulation of CYP450 enzymes has not been reported. The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g., IL-1, IL-6, IL-10, TNFα, IFN [interferon]) during chronic inflammation. Thus, as an antagonist of IL-17A, secukinumab could normalize the formation of CYP450 enzymes. Therefore, upon initiation or discontinuation of secukinumab in patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index (NTI), it may be necessary to monitor for therapeutic effect or drug concentration and consider dosage modification of the CYP450 substrate.
Dosage and Administration1,2: Secukinumab is supplied in cartons containing one or two single-use 150-mg prefilled Sensoready pens or syringes, or one 150-mg single-use vial of lyophilized powder for reconstitution. The recommended dose is 300 mg by SC injection at weeks 0, 1, 2, 3, and 4 followed by 300 mg every 4 weeks. Each 300 mg dose is given as two SC injections of 150 mg. For some patients, a dose of 150 mg may be sufficient. Patients may self-inject with prefilled pens and syringes after proper training in SC injection techniques. The lyophilized powder should be reconstituted with Sterile Water for Injection by a healthcare provider.
Dulaglutide (Trulicity, Eli Lilly and Company)
Indication and Clinical Profile3,4: Dulaglutide is a new antihyperglycemic medication approved as an adjunct to diet and exercise to improve glycemic control for adults with type 2 diabetes mellitus (T2DM). It may be used either as monotherapy for these patients or in combination with other medications such as metformin, sulfonylureas, thiazolidinediones, and insulin. T2DM is a chronic disease affecting 29 million people that is characterized by insulin resistance resulting in elevations of blood glucose levels. Patients with long-term uncontrolled T2DM are at increased risk of multiple macrovascular and microvascular complications including heart disease, vision and hearing loss, neuropathy, and nephropathy. Dulaglutide is the fourth glucagon-like peptide-1 (GLP-1) receptor agonist to receive FDA approval, the others being exenatide, liraglutide, and albiglutide.
FDA approval of dulaglutide was based on six randomized, double-blind, 52-week, clinical trials involving 3,342 patients. The trials studied dulaglutide at doses of 0.75 mg and 1.5 mg as monotherapy and in combination with metformin, metformin and glimepiride, metformin and pioglitazone, and prandial insulin with or without metformin. In all trials, dulaglutide was proven efficacious in improving blood glucose control as demonstrated by reductions in A1C from baseline.
Pharmacology and Pharmacokinetics3,4: Dulaglutide functions as a human GLP-1 receptor agonist. GLP-1 receptors are pancreatic beta-cell receptors whose endogenous ligand is normally released from gastrointestinal (GI) cells in response to food. Upon binding, GLP-1 receptors activate coupled adenylyl cyclase, resulting in an increase in intracellular cyclic adenosine monophosphate (cAMP). Beta-cell cAMP elevations lead to a more normalized blood glucose level via glucose-dependent insulin release, reduced glucagon secretion, delayed gastric emptying, and a promotion of satiety. Because GLP-1 receptor agonists like dulaglutide promote insulin release in a blood glucose–dependent manner, they are less likely than sulfonylureas or insulin to cause hypoglycemia.
The mean bioavailability of 0.75- and 1.5-mg doses of dulaglutide was 65% and 47%, respectively, and the drug reaches its peak plasma concentration after 24 to 72 hours. It is degraded via protein catabolism pathways to its component amino acids and has a half-life of 5 days. Dulaglutide has not demonstrated an ability to alter the metabolism of other medications.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (ARs) reported in patients receiving dulaglutide in clinical trials were nausea, vomiting, diarrhea, abdominal pain, and decreased appetite. Use of dulaglutide, like other GLP-1 agonists, was also associated with an increased incidence of pancreatitis. Patients taking dulaglutide should be monitored carefully for signs of pancreatitis, and it should be avoided in patients with a history of pancreatitis. The drug’s label carries a boxed warning for dose- and duration-related increases in the incidence of thyroid C-cell tumors, including medullary thyroid carcinoma (MTC) in rats. While it is unknown whether this effect occurs in humans, dulaglutide is contraindicated in patients with multiple endocrine neoplasia syndrome type II (MEN 2) or a personal or family history of MTC due to this risk.
Dulaglutide was approved with a Risk Evaluation and Mitigation Strategy (REMS) to increase awareness of the risk for acute pancreatitis and the potential risk of MTC. The FDA is also requiring multiple postmarketing studies for dulaglutide including a trial evaluating its use in pediatric patients; a study in rats assessing effects on sexual maturation, reproduction, and central nervous system (CNS) development and function; a 15-year MTC case registry; a trial comparing dulaglutide with insulin glargine on glycemic control in patients with moderate or severe renal impairment; and an outcomes trial to evaluate its cardiovascular risk in patients at high risk of cardiovascular disease. Dulaglutide has demonstrated adverse events in animal reproduction studies, and there have not been adequate studies in pregnant women.
Use of dulaglutide with insulin or insulin secretagogues increases the risk of hypoglycemia. Because of its effects on gastric emptying, dulaglutide may affect the absorption of oral medications taken concomitantly, and concomitant use of medications with an NTI should include careful monitoring of their levels.
Dosage and Administration3,4: Dulaglutide is supplied as 0.75 mg/0.5 mL and 1.5 mg/0.5 mL solutions in either single-dose pens or prefilled syringes for SC injection in the abdomen, thigh, or upper arm. The recommended starting dose is 0.75 mg once weekly. For additional glycemic control, doses can be increased to the maximum recommended dose of 1.5 mg once weekly. No dose adjustments are necessary for elderly patients or those with renal impairment. Dulaglutide should be used with caution in patients with hepatic impairment and avoided in patients aged <18 years, as it is unstudied in these patients. The drug should be stored away from light, in a refrigerator at 36°F to 46°F, although it can also be stored at room temperature for up to 14 days.
Naloxegol (Movantik, AstraZeneca Pharmaceuticals)
Indication and Clinical Profile5,6: Naloxegol is a mu-opioid receptor antagonist that has recently been approved specifically for the treatment of opioid-induced constipation (OIC) in adults with chronic noncancer pain. Opioids are routinely used for chronic pain management, with a common side effect of constipation due to reduced GI tract motility. OIC does not dissipate with time and will continue as long as the patient is using opioids. Patients with OIC typically need to use a laxative, as they experience difficulty having bowel movements, hard or lumpy stools, or incomplete evacuation.
FDA approval of naloxegol was based on two randomized, controlled trials in 1,352 participants who had OIC while taking an opioid for a minimum of 4 weeks for noncancer pain. Participants were randomly assigned to receive 12.5 or 25 mg of naloxegol or placebo once daily for 12 weeks. The primary endpoint of both studies was that patients respond to therapy, defined as ≥3 spontaneous bowel movements (SBMs) per week and an increase of ≥1 SBM per week from baseline for at least 9 out of the 12 weeks and 3 out of the last 4 weeks. For the primary outcome, both studies showed naloxegol 25 mg to be statistically superior to placebo with 44% and 40% of patients responding to naloxegol 25 mg versus a 29% response in patients taking placebo. Statistical significance for the 12.5 mg naloxegol group versus placebo was observed in Study 1 (41% vs. 29%) but not in Study 2 (36% vs. 29%).
Pharmacology and Pharmacokinetics5,6: Naloxegol is a conjugate of the mu-opioid receptor antagonist naloxone and a polyethylene glycol (PEG) polymer (FIGURE 1). The addition of the PEG polymer limits the ability of the drug to penetrate the blood brain barrier, making naloxegol selective for peripheral opioid receptors. Antagonism of mu-opioid receptors in the GI tract blocks the constipating effects elicited by opioid use.
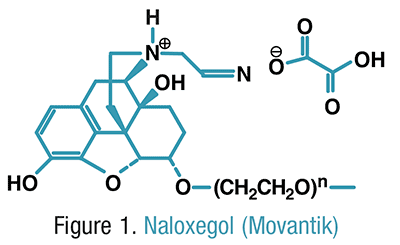
Naloxegol’s extent of absorption is affected by food, with its maximum plasma concentration (Cmax) and area under the curve (AUC) increased by 30% and 45%, respectively, when taken with a high-fat meal versus on an empty stomach. On an empty stomach, naloxegol reaches its Cmax in <2 hours and is 4.2%- bound to plasma proteins. Naloxegol is primarily metabolized via CYP3A4-mediated N-dealkylation, O-demethylation, oxidation, and partial loss of the PEG chain to six metabolites with unknown activity. Naloxegol does not appear to significantly alter the activity of any major CYP450 isozyme. The half-life is between 6 and 11 hours, and the drug is excreted primarily through feces (68%) with some renal (16%) elimination.
Adverse Reactions and Drug Interactions5,6: The most common ARs reported by patients receiving naloxegol in clinical trials included abdominal pain, nausea, vomiting, diarrhea, flatulence, and headache. Some patients experienced signs and symptoms of opioid withdrawal. Naloxegol may increase the risk of GI perforation, and its use is contraindicated in patients with known or suspected GI obstruction or at increased risk of recurrent obstruction. It should also be used with caution in patients with reduced GI tract-wall integrity. The FDA is requiring a postmarketing study of naloxegol to further evaluate potential risks of cardiovascular adverse events. Naloxegol should not be used during pregnancy unless the potential benefit to the patient offsets the potential risk to the fetus (Pregnancy Category C), as exposure during pregnancy may potentiate opioid withdrawal in the fetus.
Drug interactions with naloxegol are limited to substances that impact CYP3A4. Use of naloxegol with CYP3A4 inhibitors will result in increased exposure to naloxegol and increased risk of ARs. Patients taking moderate CYP3A4 inhibitors with naloxegol should start at a reduced dose (12.5 mg); naloxegol is contraindicated in patients taking strong CYP3A4 inhibitors. Consumption of grapefruit or grapefruit juice while on naloxegol should also be avoided. Concomitant use of strong CYP3A4 inducers is not recommended, as this may result in subtherapeutic exposure to naloxegol.
Dosage and Administration5,6: Naloxegol is supplied as a 12.5- and 25-mg tablet for oral administration, and the recommended starting dose is 25 mg once daily. For patients unable to tolerate 25 mg once daily, the dose should be reduced to 12.5 mg once daily. Prior to starting naloxegol, all other laxative therapies should be stopped and only restarted as needed if patients receive a suboptimal response from naloxegol after 3 days. No dose adjustments are recommended for elderly patients or patients with mild-to-moderate hepatic impairment. Naloxegol has not been studied in patients with severe hepatic impairment. Patients with creatinine clearance (CrCl) <60 mL/min should use an initial dose of 12.5 mg once daily. Patients should be counseled to take naloxegol on an empty stomach either 1 hour before or 2 hours after meals and to discontinue treatment if their opioid pain medication is also discontinued.
Ledipasvir-Sofosbuvir (Harvoni, Gilead)
Indication and Clinical Profile7,8: Harvoni is a combination of sofosbuvir, a previously approved hepatitis C virus (HCV) drug, and the new drug ledipasvir. It is indicated to treat chronic HCV genotype 1 (HCV-1) infection. HCV infection causes hepatic inflammation that, over time, results in diminished liver function or failure. The disease is typically asymptomatic for years until liver scarring and cirrhosis develops, causing complications such as bleeding, jaundice, ascites, infections, and liver cancer. Approximately 3.2 million Americans are infected with HCV, and 70% to 80% of cases are HCV-1 infections. Harvoni is the third drug approved by the FDA to treat chronic HCV, the others being simeprevir (Olysio) in November 2013 and sofosbuvir (Sovaldi) in December 2013. It received breakthrough-therapy designation to gain FDA approval and is the first approved HCV regimen not requiring administration with interferon or ribavirin.
Approval of Harvoni was based on three randomized phase III trials in 1,518 subjects with HCV-1 with compensated liver disease. The primary endpoint of all trials was sustained virologic response (SVR), defined as HCV RNA less than a lower limit of quantification (LLOQ) of 25 IU/mL at 12 weeks after the cessation of treatment. The first trial involved noncirrhotic, treatment-naïve participants in which SVR was achieved in 94% of those treated for 8 weeks and in 96% of those treated for 12 weeks. The second trial included both cirrhotic and noncirrhotic treatment-naïve participants where SVR was achieved in 99% of patients treated for 12 weeks. Harvoni’s efficacy in treatment-experienced participants with and without cirrhosis was demonstrated in a third trial in which 94% of patients treated for 12 weeks and 99% of those treated for 24 weeks achieved SVR. Ribavirin did not increase response rates in any trial.
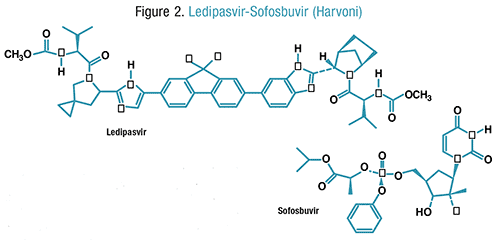
Pharmacology and Pharmacokinetics7,8: Ledipasvir, a highly functionalized fluorene, is an inhibitor of HCV NS5A protein, which is required for viral replication. Sofosbuvir is a prodrug that is intracellularly converted to an active uridine analogue triphosphate (FIGURE 2). This active metabolite functions as a chain terminator to viral RNA once it is incorporated into HCV RNA by NS5B polymerase. Sofosbuvir is a potent inhibitor of all HCV genotypes (1-6), including strains resistant to protease inhibitors. The actions of both ledipasvir and sofosbuvir result in HCV that is unable to multiply.
Ledipasvir achieves its peak plasma concentration at 4 to 4.5 hours and is 99.8% plasma protein–bound. It is metabolized via an unknown oxidative mechanism and does not appear to induce or inhibit metabolism by any major CYP isozyme. Ledipasvir is eliminated primarily in the feces (~86%) with only ~1% of the drug eliminated renally. Its half-life is 47 hours.
Sofosbuvir achieves its peak plasma concentration between 0.8 and 1 hours and is about 60% plasma protein–bound. It is converted to its active metabolite GS-461203 via hydrolysis and phosphoramidate cleavage followed by intracellular phosphorylation by the pyrimidine nucleotide biosynthetic pathway. It is inactivated via dephosphorylation to its metabolite GS-331007, which cannot be efficiently rephosphorylated. Sofosbuvir is excreted primarily in the urine (80%) mainly as its GS-331007 metabolite and has a half-life of 0.5 hours.
Adverse Reactions and Drug Interactions7,8: The most common adverse effects reported in Harvoni-treated clinical trial patients were headache, fatigue, nausea, diarrhea, and insomnia. Some patients also experienced laboratory abnormalities including elevations of bilirubin, lipase, and creatine kinase. No adverse effects were noted in animal reproduction studies of Harvoni; however, because there are no adequate studies with this drug in pregnant women, it should only be used during pregnancy if the potential benefit outweighs the potential risk to the fetus (Pregnancy Category B).
Ledipasvir’s absorption is pH-dependent; thus, use of antacids within 4 hours of Harvoni administration should be avoided. However, proton pump inhibitors and H2-receptor antagonists can be taken simultaneously or 12 hours apart from Harvoni at a dose that does not exceed doses comparable to famotidine 40 mg twice daily. Both drugs in Harvoni are substrates of the transporter P-glycoprotein (Pgp) and coadministration with Pgp inducers and inhibitors is not recommended, as they can decrease and increase serum concentrations of Harvoni. Ledipasvir is also an inhibitor of Pgp and breast cancer resistance protein (BCRP) and may increase absorption of coadministered substrates for these transporters. Harvoni should not be taken concurrently with amiodarone due to an increased risk of symptomatic bradycardia and fatal cardiac arrest via an unknown mechanism.
Dosage and Administration7,8: Harvoni is supplied as a tablet of a fixed-dose combination of ledipasvir 90 mg and sofosbuvir 400 mg. The recommended dose is one tablet once daily with or without food. No dosage adjustment is recommended for patients who are elderly, have hepatic impairment, or have CrCl >30 mL/min/1.73 m2. Harvoni was not studied in patients with CrCl <30 mL/min/1.73 m2 or those with end-stage renal disease. Duration of therapy is dependent on patient characteristics. Patients who are treatment-naïve should receive 12 weeks of therapy unless they are without cirrhosis and have a baseline HCV RNA <6 million IU/mL, in which case they should receive 8 weeks of therapy. Treatment-experienced patients without cirrhosis should take Harvoni for 12 weeks, while those with cirrhosis should receive 24 weeks of therapy.
REFERENCES
1. Cosentyx (secukinumab injection) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation; January 2015.
2. Langley RG. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326-338.
3. Trulicity (dulaglutide injection) package insert. Indianapolis, IN: Eli Lilly and Company; March 2015.
4. Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z. Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5). Diabetes Care. 2014;37(8):2149-2158.
5. Movantik (naloxegol tablets) package insert. Wilmington, DE: AstraZeneca Pharmaceuticals LP; January 2015.
6. Webster L, Chey WD, Tack J, et al. Randomised clinical trial: the long-term safety and tolerability of naloxegol in patients with pain and opioid-induced constipation. Aliment Pharmacol Ther. 2014;40(7):771-779.
7. Harvoni (ledipasvir and sofosbuvir tablets) package insert. Foster City, CA: Gilead Sciences, Inc; March 2015.
8. Kowdley KV, Gordon SC, Reddy KR, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879-1888.
To comment on this article, contact rdavidson@uspharmacist.com.



