US Pharm. 2012;37(5):HS-2-HS-7.
Pain is the most common reason that people seek medical attention.1 A recent study estimates that pain affects tens of millions of Americans and costs the United States $635 billion annually because of increased consumption of medical care and indirect costs from missed workdays and loss of productivity.2 Management of chronic pain with medications such as opioids is a common strategy, but chronic pain can persist or worsen despite aggressive opioid therapy. This article aims to shed light on the phenomenon of opioids that are prescribed to treat pain but cause new or paradoxically worsening pain. This condition is called opioid-induced hyperalgesia (OIH). OIH may be more formally defined as increased nociceptive sensitization caused by exposure to opioids.3 OIH differs distinctly from tolerance, addiction, dependence, and disease progression. The clinical prevalence of OIH is unknown.4
Diagnosis and Presentation
One of the chief problems in properly diagnosing OIH is the condition’s close resemblance to opioid tolerance. Tolerance and OIH share the characteristic of reduced analgesic response to the opioid dose, and they likely share many of the same cellular mechanisms.5 Tolerance can be overcome by increasing the opioid dose, whereas the same increase in a patient with OIH results in worsened pain.4 Additionally, tolerance tends to develop slowly over time, whereas the increased pain resulting from opioid treatment in the OIH patient occurs relatively quickly. Often, OIH differs from tolerance in that the pain intensity is stronger than initially reported.6
Undertreated pain is another possibility that should be ruled out. If opioid therapy is suboptimal, increasing the dose should lead to pain relief; if OIH is present, the opposite should be true. For an accurate diagnosis of OIH, the pain must resolve once treatment with the offending opioid is discontinued.6 To complicate matters further, hyperalgesia resulting from opioid withdrawal is a well-documented phenomenon.7 As such, pain resolution from opioid discontinuation in OIH will not be immediate and will require patience. This poses its own challenges for both clinician and patient.
OIH tends to present with other distinct characteristics (TABLE 1). Pain associated with OIH tends to be more diffuse and of lesser quality, and noxious stimuli tend to be more painful that would normally be expected. In OIH, pain often manifests in areas extending beyond the region of injury or tissue damage. Pain can persist in OIH despite removal of the original source of pain or healing of the damaged tissue. In OIH, as opioid treatment progresses, the pain may give the illusion of getting worse than originally reported despite time, rest, and other measures that would normally allow for a clinically relevant amount of healing.5 Additionally, allodynia has been demonstrated in a number of human and animal studies of OIH.8

Etiology
There are a number of theories regarding the cause of OIH. The theory that has been the most researched and is currently receiving the most attention is the neuroexcitatory model. It is believed that certain opioids and their metabolites agonize the N-methyl-D-aspartate (NMDA) receptor. Activation of the NMDA receptor causes an influx of calcium that greatly enhances the excitability of the neuron. When the NMDA receptor and corresponding neurons are more active, they can more readily transmit painful impulses initiated by circulating substance P or other noxious stimuli. Glutamate is the primary endogenous agonist of the NDMA receptor.9
Supporting this theory of pronociception modulated by NMDA receptor activation are studies showing relief of OIH after administration of NMDA receptor antagonists. These antagonists noncompetitively bind to the phencyclidine site on the NMDA channel and block the influx of calcium that would occur when the receptor binds glutamate or another agonist.10 Animal and human studies have demonstrated that subjects with OIH who were administered ketamine, an NMDA receptor antagonist, scored better during controlled pain-stimulus testing. In one study, rats were given intrathecal morphine injections for several days until they displayed OIH that was sensitive to heat. Administration of dizocilpine, an NMDA inhibitor, was effective in at least partially reversing thermal hyperalgesia.5 Other NMDA receptor antagonists, such as dextromethorphan, have demonstrated some evidence of relieving hyperalgesic states through inhibition at the NMDA receptor.
Opioids, such as codeine, hydromorphone, and morphine, undergo a number of biotransformations as part of the normal metabolic process. Codeine is metabolized into morphine by the CYP2D6 enzyme. Morphine is metabolized into a number of molecules, but it is metabolized primarily through glucuronidation into morphine-3-glucuronide (M3G) and, to a lesser extent, morphine-6-glucuronide (M6G) (FIGURE 1).11
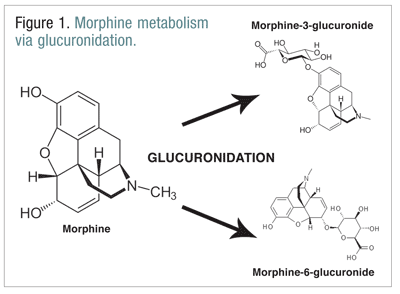
M6G is a therapeutically important metabolite. It has mu receptor binding affinity similar to that of morphine and is capable of producing analgesia, respiratory depression, and other effects similar to those of morphine. M3G, however, is not an agonist at the mu receptor, and it has little affinity for the mu receptor compared with morphine. Unlike morphine, M6G, and other opioids, M3G is an NMDA agonist. Through glucuronidation, M3G is converted at a rate about six times greater than that of M6G.12 Other opioids have been implicated in having NMDA agonist activity and paradoxical neuroexcitatory effects.6
Supporting the theory that OIH is caused by activity of receptors besides the mu opioid receptor is evidence that OIH is not alleviated by administration of an opioid antagonist (e.g., naloxone). In patients experiencing OIH while on high-dose opioid therapy, administration of naloxone caused a worsening of pain.13 By displacing the opioid from the mu receptor, naloxone prevented the opioid from providing what analgesia was possible through mu receptor activation, while concurrently the hyperalgesia now remained unchallenged.14
Dynorphin may also be involved in the development of OIH.15 Dynorphin is an endogenous opioid peptide that binds primarily to the kappa opioid receptor and, to a lesser extent, the mu opioid and NMDA receptors. It is believed that dynorphin’s agonism of the kappa opioid receptor and NMDA receptor has a role in the pronociception of OIH despite the presence of pure mu receptor agonists. Studies have shown that reversal of dynorphin can restore the analgesic effects of morphine when subjects are given a dynorphin antiserum. Furthermore, dynorphin is known to increase during prolonged opioid administration. Dynorphin also causes the release of excitatory neuropeptides within different locations in the central nervous system.
Treatment Options
Once the diagnosis of OIH has been made, there are a number of treatment options from which to carefully choose. Provided that the initial painful injury or tissue damage has resolved and the pain persists in spite of—and because of—opioid treatment, the most straightforward approach is to discontinue the offending opioid. This should be done gradually to minimize adverse withdrawal effects. It should be noted that hyperalgesia may likely worsen early in the discontinuation process.16 This presents a challenging ethical situation in which the clinician may have difficulty convincing the patient that the medication prescribed to treat pain may have been causing or worsening the pain and that the pain may get worse still before it ultimately resolves.17
If legitimate pain persists and some amount of analgesia with opioids is required, other strategies beyond total opioid discontinuation should be explored. Patients experiencing OIH may obtain relief by reducing the opioid dose.4 There are reports of patients finding an acceptable balance of analgesia and relief from hyperalgesia upon opioid dose reduction.
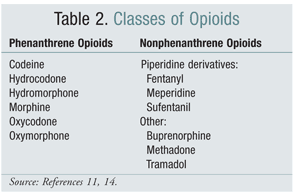
Switching from one structural class of opioids to another has been an effective option for mitigating OIH in some studies. Studies to date have demonstrated that OIH is more strongly associated with opioids from the phenanthrene class (TABLE 2).4 Titration of the phenanthrene opioid and conversion to another may provide resolution of OIH. Codeine, hydromorphone, morphine, and structurally similar opioids undergo glucuronidation as part of their metabolism. Avoidance of an NMDA receptor–activating glucuronide metabolite is possible by switching to an opioid that is structurally unique, such as fentanyl. At this writing, OIH has not been demonstrated in trials involving oxymorphone.4
Supplementing opioid therapy with a cyclo-oxygenase 2 (COX-2) inhibitor is another strategy that has some support. By reducing prostaglandin synthesis, COX-2 inhibitors can decrease the sensitization of pronociceptive neurons. Nonsteroidal anti-inflammatory drugs (NSAIDs) and COX-2 inhibitors appear to have analgesic effects independent of their ability to suppress prostaglandin synthesis peripherally.9 COX-2 inhibitors also have demonstrated the ability to antagonize the NMDA receptor. Centrally, NSAIDs are capable of antagonizing the NMDA receptor by blocking glutamate, substance P, and other excitatory amino acids. Independent of this, NMDA receptor activation can upregulate COX-2 expression.18
Given the attention the NMDA receptor has received for its role in OIH, antagonizing this receptor would seem to be a reasonable treatment strategy. Ketamine has been much researched, but its adverse effects can be severe. While it antagonizes NMDA and provides an anesthetic effect that would promote analgesia, side effects are common, diverse, and severe.19 These effects include tachyarrhythmia, hypertension, cognitive impairments, and psychomimetic reactions, including mood changes, vivid dreams, delirium, hallucinations, and sedation. Ketamine is effective in reversing hyperalgesia and augmenting the effects of opioids in patients receiving large doses, but its adverse effects prevent it from being a viable treatment option.
Dextromethorphan is another NMDA receptor antagonist that has been investigated for the treatment of OIH. A combination product containing a 1:1 mixture of morphine and dextromethorphan has been studied.20 Studies failed to demonstrate that combination therapy could achieve superior analgesia compared with morphine alone. Similar therapy failures occurred in a study that used a 2:1 mixture of the same agents. The studies have been criticized for having an insufficient amount of dextromethorphan to adequately antagonize the NMDA receptor.21 What remains unknown is the dose of dextromethorphan necessary to either augment an opioid’s analgesia or prevent the OIH caused by NMDA receptor activation.
Methadone is an opioid with unique qualities that make it a compelling option for treating pain in the patient with OIH. In addition to its analgesic effects through binding to the mu opioid receptor, methadone is a weak NMDA receptor antagonist.4 It has been reported that the addition of even a low dose of methadone has been effective for reducing hyperalgesia. In one case, the addition of low-dose methadone (10 mg twice daily) improved reports of pain markedly, with a reduction in total opioid dose of 40% to 50%.22 Switching from the offending opioid to methadone has been a popular treatment strategy. Tramadol and meperidine also have some NMDA receptor antagonist activity.
Another treatment modality that has some potential for treating OIH is buprenorphine.23,24 Buprenorphine is a partial opioid agonist at the mu receptor and also has kappa receptor antagonist activity.4 Dynorphin, a kappa receptor agonist, is known to increase during treatment with opioids. Activation of the kappa receptor largely antagonizes the mu receptor–mediated effects of opioids.25 The clinical utility of buprenorphine in treating OIH has yet to be fully explained.
Conclusion: Looking Forward
Much work remains to be done before it can be declared that OIH is understood. The exact cellular mechanism and signaling pathways responsible for this phenomenon are not defined. To complicate this, there are conflicting reports as to whether specific components play a key role, or any role whatsoever. The bulk of research on elucidating the pathophysiology of OIH has involved the animal model. This has some value, but information from the human model is scarce.
A weak understanding of the cause of a condition generates an uncertain armamentarium of treatment options. The best strategy available is total discontinuation of the problematic opioid, but this is of little use to the patient who requires opioid-type pain relief. Several other treatment modalities have been explored, but all of them would benefit from further study. The majority of studies done in humans involve perisurgical patients, recovering drug addicts, and experimental pain models. These studies are not without value, but these populations do not mirror the chronic pain population. Other limitations of available treatment options include lackluster results, excessive toxicity despite moderate success, and small studies producing weak conclusions.
At this writing, a number of clinical studies using the NMDA receptor antagonist memantine to treat OIH are ongoing or planned.26 Still, there are conflicting reports as to the exact role of NMDA receptors in OIH. Further research must be done to determine the key responsible receptors and mechanisms present in OIH. Expanding the sample size of studies will help support conclusions already made about which treatment agents are responsible for OIH and effective for treating it. Conducting studies that focus on chronic pain, rather than models of acute pain, can bring clinically relevant information to the practitioner.
Chapman and colleagues have raised specific questions that must be answered before diagnosis of OIH and effective treatment can be delivered.27 What are the relationships of opioid dose and duration to OIH? With what type of pain, or in what type of patient, is OIH more apt to develop? These are the questions that must guide future clinical research.
REFERENCES
1. Parselis Kelly J, Cook SF, Kaufman DW, et al. Prevalence and characteristics of opioid use in the US adult population. Pain. 2008;138:507-513.
2. Institute of Medicine. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: The National Academies Press; 2011.
3. Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24:479-496.
4. Lee M, Silverman SM, Hansen H, et al. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14:145-161.
5. Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100:213-217.
6. Raffa RB, Pergolizzi JV Jr. Opioid-induced hyperalgesia: is it clinically relevant for the treatment of pain patients? Pain Manage Nurs. 2011;1-17.
7. Li X, Clark JD. Hyperalgesia during opioid abstinence: mediation by glutamate and substance P. Anesth Analg. 2002;95:979-984.
8. Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain
perception: new evidence from a study of chronic opioid addicts and
healthy subjects. Drug Alcohol Depend. 2006;82:218-223.
9. Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate
or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276 -1279.
10. Fisher K, Coderre TJ, Hagen NA. Targeting the
N-methyl-D-aspartate receptor for chronic pain management. Preclinical
animal studies, recent clinical experience and future research
directions. J Pain Symptom Manage. 2000;20:358-373.
11. Lötsch J. Opioid metabolites. J Pain Symptom Manage. 2005;29:S10-S24.
12. Skarke C, Lötsch J. Morphine metabolites: clinical implications. Semin Anesth, Perioperative Med Pain. 2002;21:258-264.
13. Juni A, Klein G, Kest B. Morphine hyperalgesia in mice is
unrelated to opioid activity, analgesia, or tolerance: evidence for
multiple diverse hyperalgesic systems. Brain Res. 2006;1070:35-44.
14. Angst M, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570-587.
15. Vanderah TW, Gardell LR, Burgess SE, et al. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074-7079.
16. Hooten WM, Mantilla CB, Sandroni P, Townsend CO. Associations
between heat pain perception and opioid dose among patients with chronic
pain undergoing opioid tapering. Pain Med. 2010;11:1587-1598.
17. Silverman S. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician. 2009;12:679-684.
18. Li SQ, Xing YL, Chen WN, et al. Activation of NMDA receptor is
associated with up-regulation of COX-2 expression in the spinal dorsal
horn during nociceptive inputs in rats. Neurochem Res. 2009;34:1451-1463.
19. Okon T. Ketamine: an introduction for the pain and palliative medicine physician. Pain Physician. 2007;10:493-500.
20. Galer BS, Lee D, Ma T, et al. MorphiDex (morphine
sulfate/dextromethorphan hydrobromide combination) in the treatment of
chronic pain: three multicenter, randomized, double-blind, controlled
clinical trials fail to demonstrate enhanced opioid analgesia or
reduction in tolerance. Pain. 2005;115:284-295.
21. Wiesenfeld-Hallin Z. Combined opioid-NMDA antagonist therapies.
What advantages do they offer for the control of pain syndromes? Drugs. 1998;55;1-4.
22. Vorobeychik Y, Chen L, Bush MC, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med. 2008;9:724-727.
23. Baron MJ, McDonald PW. Significant pain reduction in chronic pain patients after detoxification from high-dose opioids. J Opioid Manag. 2006;2:277-282.
24. Koppert W, Ihmsen H, Körber N, et al. Different profiles of
buprenorphine-induced analgesia and antihyperalgesia in a human pain
model. Pain. 2005;118:15-22.
25. Pan ZZ. Mu-opposing actions of the kappa-opioid receptor. Trends Pharmacol Sci. 1998;19:94-98.
26. Ballantyne JC, Mao J. Opioid therapy for chronic pain. N Engl J Med. 2003;349:1943-1953.
27. Chapman CR, Lipschitz DL, Angst MS, et al. Opioid pharmacotherapy
for chronic non-cancer pain in the United States: a research guideline
for developing an evidence-base. J Pain. 2010;11:807-829.
To comment on this article, contact rdavidson@uspharmacist.com.


