US Pharm. 2016;41(1):35-40.
ABSTRACT: Optic neuritis (ON) is an inflammatory condition of the optic nerve. Based upon disease etiology, ON is broadly classified as typical or atypical. Typical ON is strongly associated with multiple sclerosis (MS), while atypical ON is unrelated to MS. The two types differ in etiology, pathophysiology, and treatment. Successful clinical management depends upon the correct differential diagnosis. Selected typical ON patients may benefit from short-term, high-dose corticosteroids. Disease-modifying drugs may prevent or delay progression to first occurrence or relapse of existing MS, as well as prevent another ON episode. Atypical ON may require both corticosteroids and long-term immunosuppressant therapy, depending on the etiology.
Optic neuritis (ON) is an inflammatory condition that affects the optic nerves. Typical ON is associated with multiple sclerosis (MS), while atypical ON is a manifestation of other diseases or conditions. Although many cases of typical ON resolve with full restoration of vision, some cases may progress to MS, visual abnormality, and/or vision loss.1,2 The purpose of this review is to briefly present the current under-standing of ON and its clinical management.
Classification
Based upon disease etiology, ON is classified as typical or atypical. While typical ON is strongly associated with MS, atypical ON is linked to a wide variety of disorders that are unrelated to MS (refer to TABLE 1).1-6
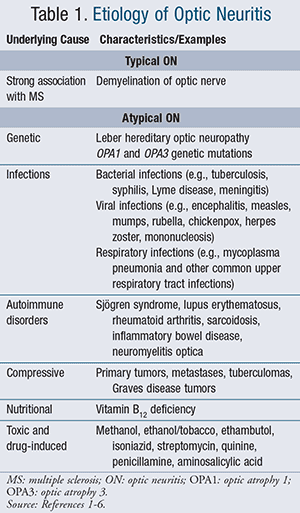
Atypical ON
Genetic Disorders: Leber hereditary optic neuropathy (LHON) is an inherited form of vision loss, which usually presents in the teens or early twenties, affecting males much more commonly than females. Genetic mutations of mitochondrial proteins may eventually lead to optic nerve cell death and a spectrum of symptoms from no vision loss to profound and permanent loss of central vision.3
Optic atrophy 1 (OPA1 ) and optic atrophy 3 (OPA3 ) genetic mutations lead to defective or missing proteins that subsequently result in mitochondrial dysfunction and deterioration of structures that transmit visual infor-mation from the eyes to the brain.4,5
Infections: A wide variety of bacterial, viral, and upper respiratory tract infections have been associated with atypical ON. Although less common etiologic agents, differential diagnosis must rule out these infectious causes of ON.6
Autoimmune Disorders: ON may occur in systemic autoimmune diseases such as Sjögren syndrome, lupus erythematosus, rheumatoid arthritis, sarcoidosis, and inflammatory bowel disease. In these conditions, the immune system mistakenly targets the myelin sheath covering the optic nerve, and in some cases, other ocular and neurologic structures.7
Commonly associated with ON, neuromyelitis optica (NMO) is characterized by inflammatory lesions in the optic nerves and the spinal cord. The presence of Aquaporin-4 (AQP4) antibodies, a biomarker for NMO, is very helpful in establishing a diagnosis of NMO. Although similar to MS, NMO less often involves damage to nerves in the brain. The course of the ON disease process is more severe as compared to MS, with over 50% of patients becoming blind or wheelchair dependent after 5 years.7,8
Compressive Tumors: Any tumor that compresses structures relaying visual messages from the retina to the brain may mimic signs and symptoms of ON. Differential diagnosis should eliminate these possible pathologies.9
Nutritional: While rare, vitamin B12 deficiencies have been associated with ON. This form of ON is reversible and treatable. Malnourished patients who present with ON symptomatology, and no other identifiable risk factors, should be evaluated for this deficiency.1,10
Toxic and Drug-induced: Several chemicals and drugs can elicit signs and symptoms that mimic ON (TABLE 1). These should be considered in the differential diagnosis of ON. The most important aspect of treatment and potential restoration of vision is identification and prompt withdrawal of the offending agent.11
Prevalence and Risk Factors
Geographically, typical ON occurs more frequently among populations dwelling in regions of higher latitude than among those who reside close to the equator.12 The incidence of typical ON is believed to be lower in Asian countries than in the United States.13 The incidence of atypical ON is dependent on disease etiology. In general, atypical ON is less prevalent in regions where typical ON is prominent.1
The risk factors for typical and atypical ON include age, gender, ethnicity, and genetic factors. Typical ON is highly prevalent among those 20 to 55 years of age, and is usually unilateral.1,14 However, it is rare in the pediatric population.15 Typical ON is more prevalent in women than men. Some studies have shown that prevalence among Caucasians is higher than that among Blacks.1 Finally, some genetic mutations increase the risk of typical ON.1 In contrast to typical ON, atypical ON is highly prevalent in pediatric patients and adults over 50 years of age, as well as Black, Asian, and Polynesian populations.1 TABLE 2 provides a summary of risk factors associated with typical and atypical ON.1
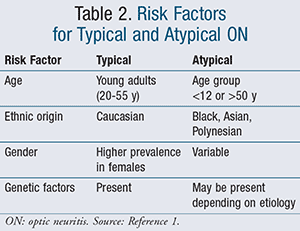
Anatomy and Physiology
Axonal retinal ganglion cells converge at the optic disc and transition into the optic nerves. The optic nerve for each eye crisscrosses at the optic chiasm to the opposite side of the brain. At this point, the optic nerves become the optic tracts. The optic tracts terminate at visual centers in the brain, with the right brain controlling the left eye and the left brain controlling the right eye (FIGURE 1).9 The optic chiasm is located directly above the pituitary gland, accounting for vision problems that may be induced by pituitary gland tumors pressing on the optic chiasm.9
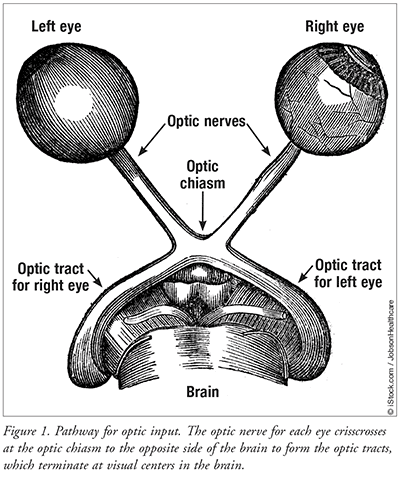
The function of the optic nerves is to transmit information from the retina to the brain for visual perception. Light striking the photoreceptor cells triggers a neurotransmitter response that generates action potential in the axons of the retinal ganglion cells for transmission to the brain via the optic nerves. The myelin sheath accelerates the conduction of the action potential through the axons of the optic nerves and tracts. Myelin is present in segments along the axons (FIGURE 2). This arrangement speeds up conduction of nerve impulses as the action potential jumps from segment to segment along the axons to visual centers in the brain.9
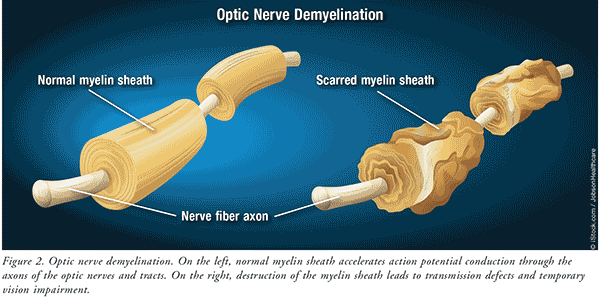
Pathophysiology
Several conditions that interfere with optic nerve function may cause ON. Regardless of etiology, the final end point of ON involves axon demyelination, axon damage, or both.
The primary pathogenicity in typical ON is optic nerve demyelination associated with inflammation.16,17 Pathologic changes involving various retinal structures may precede this occurrence.17 Destruction of the myelin leads to transmission defects with consequent temporary vision impairment (FIGURE 2).14,18 Vision loss due to demyelination is usually temporary because the myelin can be regenerated. Unfortunately, some cases involve more severe inflammation accompanied by release of T and B lymphocytic cells and cytokines that cause axonal damage and possible permanent vision loss.14,18
The thickness of the retinal nerve fiber layer (RNFL) is increased during the acute stage of ON, implying the occurrence of optic nerve swelling. Conversely, after an ON attack, the RNFL is significantly reduced.1,19 A thinner than normal layer indicates axonal loss.1,2
While the final end point of ON is the same irrespective of etiology, the triggers and resultant processes that lead to these outcomes vary widely depending upon the underlying cause.1,2,8 For example, some infectious causes of ON may be characterized by increased white blood cells, elevated protein concentration, and low glucose concentrations in the cerebrospinal fluid (CSF), which are rarely found in cases of typical ON.1
Clinical Signs and Symptoms
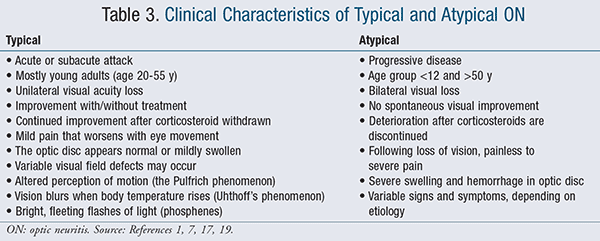
Pharmacists should be aware of some of the common symptoms associated with ON, as they can facilitate early detection and mitigate lasting damage to the eyes. The clinical signs and symptoms associated with ON include loss of peripheral or central vision, orbital pain that aggravates with eye movement, loss of color vision, and less commonly, phosphenes, a term that refers to bright, rapid flashes of light associated with eye move-ments.14 Other signs include loss of visual acuity, visual field defects, reduction in vision contrast and sensitivity, pupillary abnormality, and fundus abnormality.14,17 The clinical characteristics of typical and atypical ON are summarized in TABLE 3.1,7,17,19
Diagnostic Tests and Monitoring
Because many diseases may lead to ON, it is very important for the clinician to consider the differential diagnoses to ensure proper clinical management. In general, the diagnosis, treatment, long-term management, and prognosis of ON are dependent on the etiology of the disease.2 Several tools are utilized to diagnose and monitor ON, irrespective of etiology, including MRI, the visual evoked potential (VEP), and optical coherence tomography (OCT).1,14,17 MRI is used to diagnose ON and monitor its status during treatment. It is further employed to assess the risk of ON converting to MS.20 VEP assesses the alteration in visual acuity that occurs due to demyelination. OCT is used to measure the thickness of the RNFL and determine the extent of axonal atrophy.1,14,17
Medical follow-up for typical ON includes visual acuity monitoring and visual field testing upon onset and periodically thereafter, as well as referral to an MS specialist to monitor for progression to, or management of, existing MS.1 In 15% to 20% of patients, ON is the initial symptom of MS, and 50% of MS patients will experience an episode of ON at some time during the course of their disease.12,18
Treatment
Successful treatment depends upon accurately diagnosing ON, while etiology will dictate appropriate therapy goals and drug selection as discussed below.
Typical ON
In the short term, the goals of treatment for typical ON are to halt the course of inflammation, speed up the recovery process, and improve vision. Long-term goals are to prevent recurrence of ON and preclude, delay, or ameliorate a subsequent initial MS episode.12,19
There is an established role for high-dose IV corticosteroid therapy in the treatment of typical ON. Low-dose corticosteroids, whether used as stand-alone therapy or as a taper to high-dose parenteral therapy, are contraindicated because of an increased risk for ON relapsing.2
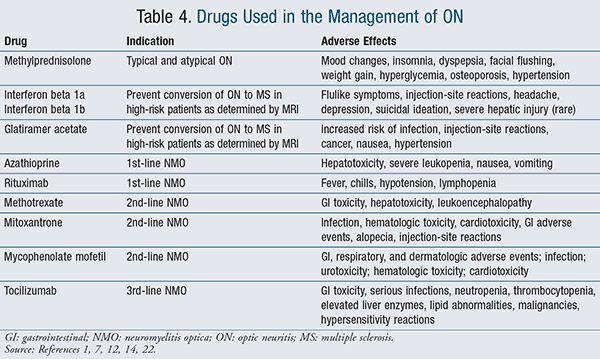
While some studies have demonstrated that high-dose parenteral corticosteroid therapy may hasten vision improvement, other studies have cast doubt on this conclusion.21 High-dose, short-term IV corticosteroid therapy is associated with few adverse effects12,19; thus, it may be worth trying this therapy in selected patients experiencing acute typical ON. Patients who may benefit from this intervention include those who require rapid vision recovery, such as patients who only have vision in one eye, severe vision loss in both eyes, or occupations that require normal visual acuity.12,18 A typical dosing regimen is IV methylprednisolone 1 g daily for 3 days (refer to TABLE 4 for indications and adverse effects).1,2
It has been definitively established that regardless of corticosteroid use, there is no change in the long-term visual outcomes of ON. Thus, typical ON is also known as steroid-independent ON because vision can improve without corticosteroid treatment.2
Patients with ON and abnormal brain MRIs may be treated with interferon beta or glatiramer acetate, which are disease-modifying drugs (DMDs), to expand the intervals between episodes, decrease the number of demyelinating attacks, and slow down disease progression (TABLE 4).12
Atypical ON
Because atypical ON has myriad causes, treatment centers on the underlying pathology. Identification and treatment, or eradication, of infections and compressive tumors, and other etiologies such as nutritional, toxic, and drug-induced causes, should be pursued. Immunosuppressive treatment specific to each autoimmune disease is warranted.7
Based upon Neuromyelitis Optica Study Group treatment guidelines, various DMDs are indicated for NMO. Azathioprine or rituximab are considered first-line agents, while methotrexate, mitoxantrone, mycophenolate, and tocilizumab are alternative second- or third-line choices (TABLE 4).7,22
Long-term corticosteroid treatment is necessary with some of the atypical ON etiologies to prevent relapse.2 It is pertinent to note that there are no established, definitive treatment guidelines for atypical ON. Evidence is mostly retrospective, observational, and not derived from randomized, controlled clinical studies. This constitutes mainly level IV evidence for clinical studies, on a scale of I to V, with level I being the strongest evidence and level V being the weakest.23 There are no consensus treatment guidelines; rather, treatment patterns appear to vary based upon local custom.1 TABLE 4 lists some of the more common agents that pharmacists may see used in different dose-combinations based upon physician preference and patient response.
Investigational Drugs
Demyelination and axonal loss are the major reasons for vision loss or impairment. Corticosteroids and DMDs cannot reverse these pathologic changes, but they may improve visual function in the short term. Thus, research has focused on investigational drugs to stop the progression of demyelination and axonal loss, repair the existing damage, and lead to better long-term visual outcomes, but to date none have been clinically successful.1
Conclusion
Typical ON is associated with MS, and corticosteroid therapy may benefit selected patients short term. Therapeutic intervention with DMDs may prevent occurrence of a first MS attack or delay progression to a recurrent episode in existing MS. The etiology of atypical ON varies widely and dictates treatment options. NMO, an autoimmune disorder, is often linked to poor patient outcomes, with over 50% of patients becoming blind or wheelchair bound after 5 years.
REFERENCES
1. Toosy AT, Mason DF, Miller DH. Optic neuritis. Lancet Neurol. 2014;13(1):83-99.
2. Pula JH, Macdonald CJ. Current options for the treatment of optic neuritis. Clin Ophthalmol. 2012;6:1211-1223.
3. Leber hereditary optic neuropathy. Genetics Home Reference. Reviewed December 2013. http://ghr.nlm.nih.gov/condition/leber-hereditary-optic-neuropathy. Accessed October 31, 2015.
4. OPA1. Genetics Home Reference. Reviewed June 2009. http://ghr.nlm.nih.gov/gene/OPA1. Accessed October 31, 2015.
5. OPA3. Genetics Home Reference. Reviewed July 2014. http://ghr.nlm.nih.gov/gene/OPA3. Accessed November 1, 2015.
6. Optic neuritis. MedlinePlus. Updated February 23, 2015. www.nlm.nih.gov/medlineplus/ency/article/000741.htm. Accessed October 31, 2015.
7. Malik A, Ahmed M, Golnik K. Treatment options for atypical optic neuritis. Indian J Ophthalmol. 2014;62(10):982-984.
8. Petzold A, Plant GT. Diagnosis and classification of autoimmune optic neuropathy. Autoimmun Rev. 2014;13(4-5):539-545.
9. Goldberg JL. Optic nerve. In: Levin LA, Nilsson FE, Ver Hove J, et al, eds. Adler’s Physiology of the Eye. 11th ed. Elsevier Inc; 2011:550-573. www.clinicalkey.com/#!/content/book/3-s2.0-B9780323057141000285. Accessed July 29, 2015.
10. Chavala SH, Kosmorsky GS, Lee MK, Lee MS. Optic neuropathy in vitamin B12 deficiency. Eur J Intern Med. 2005;16(6):447-448.
11. Hickman SJ, Ko M, Chaudhry F, et al. Optic neuritis: an update typical and atypical optic neuritis. Neuroophthalmology. 2008;32(5):237-248.
12. Osborne B, Balcer L. Optic neuritis: prognosis and treatment. UptoDate. Updated February 20, 2015. www.uptodate.com. Accessed October 31, 2015.
13. Lau PP, Yau GS, Lee JW, et al. Optic neuritis in Hong Kong: a 1-year follow-up study. Int Ophthalmol. 2015;35(3):303-310.
14. Chan JW. Optic neuritis. In: Chan JW, ed. Optic Nerve Disorders: Diagnosis and Management. 2nd ed. New York, NY: Springer Science + Business; 2014:1-40.
15. Bonhomme GR, Mitchell EB. Treatment of pediatric optic neuritis. Curr Treat Options Neurol. 2012;14:93-102.
16. Kale N. Management of optic neuritis as a clinically first event of multiple sclerosis. Curr Opin Ophthalmol. 2012;23(6):472-476.
17. Chan JW. Early diagnosis, monitoring, and treatment of optic neuritis. Neurologist. 2012;18(1):23-31.
18. Osborne B, Balcer L. Optic neuritis: pathophysiology, clinical features, and diagnosis. UptoDate. Updated March 24, 2015. www.uptodate.com. Accessed July 29, 2015.
19. Shams PN, Plant GT. Optic neuritis: a review. Int MS J. 2009;16(3):82-89.
20. Volpe NJ. The optic neuritis treatment trial: a definitive answer and profound impact with unexpected results. Arch Ophthalmol. 2008;126(7):996-999.
21. Gal RL, Vedula SS, Beck R. Corticosteroids for treating optic neuritis. Cochrane Database Syst Rev. 2012;18;4:CD001430.
22. Optic neuritis drugs. Clinical Pharmacology [online database]. www.clinicalpharmacology.com. Accessed August 3, 2015.
23. Levels of evidence for clinical studies. Elsevier. http://cdn.elsevier.com/promis_misc/Levels_of_Evidence.pdf. Accessed December 9, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.


