US Pharm. 2020;45(1):HS-2-HS-7.
ABSTRACT: Approximately 3 million people worldwide suffer from multiple sclerosis (MS). A chronic disease of the central nervous system, MS patients suffer from multiple neurological, somatosensory, and cognitive dysfunctions. The disease may broadly be presented as alternating phases of disease progression followed by a decrease or absence in symptoms, or continuous disease progression. The exact mechanism of MS is not correctly understood, although several environmental, immunological, and genetic factors have been suggested in the pathogenesis of the disease. There is no cure for MS. However, disease progression can be effectively impeded by pharmacologic agents. Disease-modifying therapies (DMTs) are widely used to slow down the progression of MS. DMTs are available either as parenteral drugs or oral agents. In this article, up-to-date data on orally active DMTs are presented.
Approximately 2 million to 3 million people have been diagnosed with multiple sclerosis (MS) worldwide. In the United States alone, about 400,000 individuals have been reported to suffer from MS. MS is a chronic autoimmune disease of the central nervous system (CNS).1-4 The condition is three times more common in women than men. People from all ethnicities have been diagnosed with MS, with Caucasians having the highest incidence. Onset of the disease is usually seen between the ages 20 and 50 years. Nevertheless, children and elderly people may also suffer from the condition.4
The exact cause of MS is unknown, but several genetic, immunologic, and environmental factors have been proposed to cause the disease. At the cellular level, myelin sheath of the axons of the CNS neurons are affected by this condition.4 It is currently believed that peripheral inflammatory cells (lymphocytes) penetrate the CNS and “strip off” the myelin sheath (composed of layers of lipid that protect and insulate axons). This results in impaired conduction of neuronal impulses in the brain. Furthermore, demyelinated axons become susceptible to irreversible degeneration by cellular insults (e.g., from free radicals). In the disease process, inflammatory cells tend to destroy oligodendrocytes responsible for neuronal protection and biosynthesis of myelin.4
There are four types of MS: relapsing remitting MS (RRMS), secondary progressive MS (SPMS), primary progressive MS (PPMS), and progressive relapsing MS (PRMS). Approximately 90% of MS patients suffer from RRMS. This form is presented by new attack or intensifying neurological symptoms followed by periods of partial or even complete recovery (also referred to as remission).1 Patients with RRMS progress to SPMS, usually after 10 to 15 years, where disability worsens and relapse/remission phases become difficult to separate. Approximately 10% to 15% of patients suffer from PPMS, where symptoms progressively worsen from onset.1 In this form, phases of relapse or remission are virtually indistinguishable. PRMS is a rare form of the disease (approximately 5% of all MS cases). In PRMS, progression of disease is steady with well-defined relapses but there is no remission.1
Patients with MS usually present with broad-based symptoms. Common symptoms of the disease include fatigue, walking difficulties, loss of balance, numbness of the face and extremities, spasticity, weakness, blurred vision, diplopia, optic neuritis, dizziness, vertigo, constipation, bladder problems, pain, sexual dysfunction, cognitive dysfunctions (memory loss, inability to process information), and depression, along with other emotional problems.2
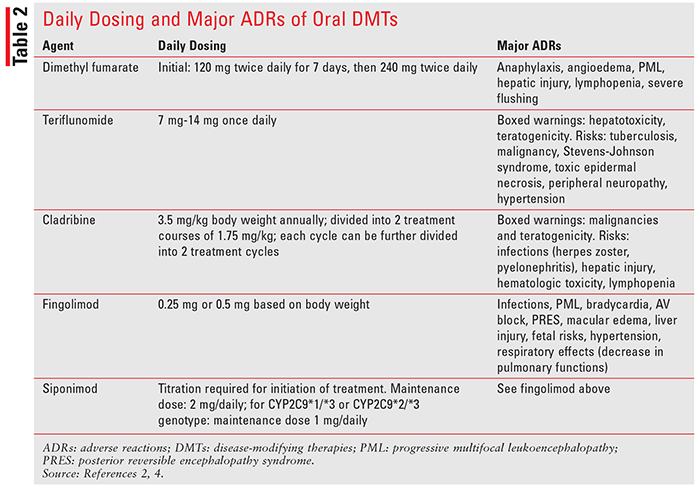
Similar to some neurological diseases, there is no cure for MS. Treatment goals include utilization of pharmacologic agents to decelerate disease progression, preserve physical activity, and suppress autoimmune reactions. In addition to relapse-management agents (IV methyl prednisolone, oral prednisolone, and IM and SC adrenocorticotropic hormone), disease-modifying therapies (DMTs) are used to alter the course of the disease and slow down associated disability. Based on routes of administration, DMTs can be broadly divided into two categories: parenteral and oral route. Parenteral-route DMTs include glatiramer acetate, different interferon formulations, natalizumab, ocrelizumab, mitoxantrone, and alemtuzumab. Oral agents include dimethyl fumarate, teriflunomide, cladribine, fingolimod, and siponimod. This article aims to provide pharmacologic properties of oral DMTs and summarize important clinical trials evaluating these agents’ efficacy and safety in MS.2-4 Mechanism of action (also in TABLE 1), adverse reactions, and pharmacokinetic properties of oral DMTs are discussed below. Daily dosing of these agents is included in TABLE 2.

Dimethyl Fumarate
Dimethyl fumarate is an ester prodrug of fumaric acid. The drug is postulated to be an activator of the erythroid-derived nuclear factor 2–like (Nrf-2) transcription pathway. This pathway is involved in cellular response to oxidative stress (e.g., increased expression of antioxidative enzymes). It also activates nicotinic acetylcholine receptors. Adverse effects of dimethyl fumarate include anaphylaxis and angioedema, flushing, lymphopenia, risk of progressive multifocal leukoencephalopathy (PML), and liver injury.4
Teriflunomide
Teriflunomide, an active metabolite of an antirheumatic drug, leflunomide, inhibits dihydroorotate dehydrogenase.4 Dihydroorotate dehydrogenase is responsible for pyrimidine synthesis, resulting in decreased lymphocyte activation and multiplication. In MS, the drug is believed to inhibit lymphocyte activation and inflammation in the CNS. The drug is not metabolized by CYP, but it inhibits CYP2C8 and induces CYP1A2. The most common adverse effects of teriflunomide include nausea, vomiting, diarrhea, alopecia, and elevated liver function tests (LFTs). Boxed warnings associated with teflunomide use include hepatotoxicity and teratogenicity.4 If needed, its excretion can be accelerated by cholestyramine and activated charcoal.
Cladribine
Cladribine is a deoxyadenosine derivative that requires phosphorylation to its triphosphate to become active. Initially, it is phosphorylated into monophosphate form by deoxycytidine kinase and eventually becomes a triphosphorylated derivative. In its triphosphate form, the drug can be incorporated into the DNA molecule to inhibit DNA synthesis and DNA repair processes.4 As an anticancer agent, cladribine has high specificity for lymphoid cells and is commonly used in hairy cell leukemia. Although it significantly decreases progression of MS, its use is associated with increased risk of malignancy. It is also a teratogenic compound capable of causing birth defects and cannot be used in pregnant women. Other major adverse reactions include lymphopenia and increased risk of infections.4
Fingolimod
Derived from a fungal metabolite myriocin, fingolimod hydrochloride is a sphingosine-1-phosphate (S1P) receptor modulator.4 The S1P is involved in the release of lymphocytes from lymphoid tissues (e.g., the thymus gland). Fingolimod is converted to fingolimod phosphate by kinases, followed by binding and activation of S1P receptors. Binding to the S1P receptor results in blocking lymphocyte migration and decreased inflammation. Major adverse effects include first-dose bradycardia, macular edema, infections, a decrease in forced expiratory volume (FEV1), elevated systolic and diastolic blood pressure, increase in LFTs, and rare occurrences of lymphopenia.4
Siponimod
Siponimod, another S1P receptor modulator, was approved in March 2019. This agent has strong affinity to S1P receptors 1 and 5 and inhibits release of lymphocytes from lymphoid organs.4 Side effects include reactivation of varicella zoster virus, elevation of liver enzymes, macular edema, and pulmonary dysfunction. Genetic testing of CYP2C9 is required prior to siponimod therapy for the following reasons: 1) this agent is contraindicated in patients with a CYP2C9*3/*3 genoytpe; 2) dose modification may be required for patients with CYP2C9*1/*3 or CYP2C9*2/*3 genotypes; and 3) siponimod metabolism will be impaired by strong inducers or inhibitors of CYP2C9 and CYP3A4.2,4
Important Clinical Trials
Dimethyl Fumarate
In a double-blind, placebo-controlled phase III trial, patients with RRMS received dimethyl fumarate orally at a dose of either 240 mg twice daily or 240 mg thrice daily or placebo for 2 years. The proportion of patients with a relapse at 2 years was 27% and 26% with twice- or thrice-daily dosing, respectively, whereas the placebo group had 47% with a relapse.5 The annualized relapse rate was significantly lower with both doses of dimethyl fumarate compared with placebo. The likely fraction of patients with confirmed progression of disability was 16%, 18%, and 26% for twice-daily dosing, thrice-daily dosing, and the placebo group, respectively. With regards to MRI, dimethyl fumarate reduced new and enhanced brain lesions.5 Adverse reactions produced by the test drug included flushing, diarrhea, nausea, and upper abdominal pain. Furthermore, lymphocyte counts were decreased and LFTs were increased.
In another trial, Fox et al compared dimethyl fumarate versus the placebo group on efficacy and safety for a 2-year period.6 Patients suffering from RRMS received an oral dose of 240 mg dimethyl fumarate twice or thrice daily versus placebo group. Glatiramer acetate (20 mg SC daily) was used as a comparator drug. Dimethyl fumarate at both doses and glatiramer acetate significantly lowered annualized response rate as well as reduced new/enlarged brain lesions.6 No effect on disability progression was observed.6 Adverse reactions of flushing, gastrointestinal events, decreased lymphocyte number (for dimethyl fumarate), and injection-site reactions (glatiramer acetate) were reported. Also, Arnold et al reported that dimethyl fumarate 240 mg twice- but not thrice-daily dosing decreased brain volume loss from 0 to 2 years by 21% compared with the placebo group.7
Teriflunomide
In the Teriflunomide Multiple Sclerosis Oral multicenter, phase III trial, teriflunomide’s ability in reduction of relapse rate and established progression of physical disability for at least 12 weeks was investigated in patients with RRMS.8 Patients were randomized into three groups (1:1:1): placebo or 7 mg or 14 mg teriflunomide once-daily orally for 108 weeks. Teriflunomide intervention reduced annualized response rate (0.54 for placebo group vs. 0.37 for teriflunomide [both 7 mg and 14 mg]). Furthermore, relative risk was significantly reduced by both teriflunomide dosages. The proportion of patients with confirmed disability progression was 22% (with 7 mg), 20% (with 14 mg), and 27% with placebo. Major adverse effects observed were elevated LFTs and serious infections (e.g., cytomegalovirus infections).8
In another randomized, double-blind, placebo-controlled, phase III trial, three groups of RRMS patients received a once-daily dose of 7 mg teriflunomide, 14 mg teriflunomide, or placebo for approximately 48 weeks (the trial was concluded at 48 weeks after inclusion of the last patient).9 Annualized response rate was much higher in the placebo group than patients on terflunomide 14 mg (P = .0001) or on teriflunomide 7 mg (P = .018). Teriflunomide 14 mg (but not 7 mg) reduced the sustained accumulation of disability score.9 Adverse effects reported in the trial include elevated LFTs, headache, serious infections (septicemia), and hair thinning.
Cladribine
Patients with RRMS (n = 1,326) in a 96-week-long, phase III, double-blind clinical trial were randomized in 1:1:1 ratio and received cladribine (either 3.5 or 5.25 mg/kg) or placebo in two or four courses during the first 48 weeks. In the second 48 weeks, each group received two courses at weeks 48 and 52 week, respectively. Cladribine-treated patients had a markedly decreased annualized relapse rate (P <.001) and higher relapse-free rate (P <.001) compared with placebo.10 Also, cladribine treatment significantly decreased 3-month sustained degree of disability and brain lesion count as detected by MRI. Frequent adverse effects observed were lymphocytopenia (21%-31%) and herpes zoster infections.10
In a 2-year extension study, efficacy and safety of cladribine was evaluated in the participants of the CLARITY trial.10,11 Of a total of 806 patients enrolled in the study, placebo recipients of CLARITY trials received 3.5 mg/kg bodyweight of cladribine.10,11 Cladribine recipients of CLARITY study were rerandomized into 2:1 receiving 3.5 mg/kg cladribine and placebo, respectively. Blinding procedure was utilized in the randomization process. Lymphopenia (grade -3) was found to be higher in the cladribine group compared with placebo. Improvement of efficacy with previous cladribine treatment (CLARITY trial) was maintained in the placebo group of the extension study (approximately 75% were found to be relapse-free). However, cladribine treatment for another 2-year period after previous cladribine therapy was not found to be efficacious.11
Fingolimod
A 2-year-long, double-blind, randomized study in 1,272 patients with RRMS received oral fingolimod at a dose of 0.5 mg or 1.25 mg daily or placebo. The primary endpoint of the study was annualized relapse rate, while the secondary endpoint was the time to disability progression.12 Out of 81% patients completing the study, annualized relapse rates were markedly reduced by either dose of fingolimod. Fingolimod at both doses significantly decreased the risk of disability progression for a 2-year period. MRI measures including new or enlarged brain lesions and brain volume loss were significantly decreased by fingolimod therapy.12 Adverse effects reported in the study were bradycardia and atrioventricular conduction block, macular edema, increased LFTs, and mild elevation of blood pressure.
Cohen et al compared fingolimod with interferon beta-1a in patients with RRMS.13 These patients received either 1.25 mg or 0.5 mg fingolimod daily or a weekly dose of 30 mcg interferon beta-1a intramuscular injection for 1 year. Of the 89% patients who completed the study, the annualized response rate was significantly lower in both fingolimod groups compared with the patients receiving interferon therapy. The findings were corroborated by MRI findings.13
Siponimod
A dose-response phase II study of siponimod recruited adult patients with RRMS. In one cohort, patients randomized at 4:4:1 ratio received an oral daily dose of siponimod 1.25 mg, 0.25 mg, or placebo for 3 months.14 A dose-response relation effect with siponimod was observed with a reduction in combined unique active brain lesions at 3 months.14 In an extension of the phase II BOLD study, RRMS patients were evaluated for a period of 24 months with different doses of siponimod to determine safety and efficacy.14,15 Siponimod was administered at 10-mg, 2-mg, 1.25-mg, 0.5-mg, and 0.25-mg daily doses.15 A total of 159 patients completed the study. Reduction in gadolinium-enhanced T1 lesion counts in the brain from the last BOLD assessment were maintained with the four highest doses for 24 months.14,15 Patients who received the three highest doses had a lower annualized relapse rate and fewer new/enlarged T2 lesion counts.15
A double-blind, phase III, randomized, controlled, multinational trial with patients suffering from SPMS Expanded Disability Status Scale (EDSS) scores of 3.0 to 6.5 (EDSS scale 0 to 10, higher value indicates greater disability) received siponimod 2 mg once daily or placebo (2:1 ratio) for up to 3 years.16 The primary endpoint was combined disability progression (CDP; defined as 1-point increase in EDSS if the baseline score was 3.0 to 5.0, or a 0.5-point increase if the baseline score was 5.5-6.5) at every 3-month evaluation. Siponimod treatment significantly reduced the risk of CDP at 3 months by 21%. Fifty-five percent reduction of annualized relapse rate was also observed in the siponimod group compared with placebo. Noteworthy adverse effects in siponimod-treated patients were lymphopenia, increased LFTs, bradycardia at treatment initiation, macular edema, hypertension, varicella zoster reactivation, and convulsions.16
Conclusion
The clinical knowledge and expertise of a pharmacist are essential for appropriate selection of a drug therapy. This becomes more of a concern when an MS patient suffers from a preexisting disease condition (e.g., MS patients with bradycardia should not receive fingolimod). Monitoring of serious toxicities associated with the use oral DMTs is equally important. Healthcare-system pharmacists, therefore, regularly assess organ functions and hematologic parameters of patients receiving oral DMTs. Associated complications of MS such as pain, bladder/bowel irregularities, and fatigue can be managed effectively with a pharmacist’s intervention as well. Lastly, but not least, pharmacists can effectively educate patients on drug therapy to ensure adherence and desired therapeutic outcome.
REFERENCES
1. Rae-Grant A, Day GS, Marrie RA, et al. Practice guideline recommendations summary: disease-modifying therapies for adults with multiple sclerosis: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90:777-788.
2. Costello K, Kalb R. The use of disease-modifying therapies in multiple sclerosis. Principles and current evidence. www.nationalmssociety.org/getmedia/5ca284d3-fc7c-4ba5-b005-ab537d495c3c/DMT_Consensus_MS_Coalition_color; Accessed September 2019.
3. Saguil A, Kane S, Farnell E. Multiple sclerosis: a primary care perspective. Am Fam Physician. 2014;90:644-652.
4. Bainbridge JL, Miravalle A, Wong PS. Multiple sclerosis. In: DiPiro JT, Talbert RL, Yee GC et al. eds. Pharmacotherapy: A Pathophysiologic Approach. 10th ed. New York, NY: McGraw-Hill Medical; 2017:815-835.
5. Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098-1107.
6. Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087-1097.
7. Arnold DL, Gold R, Kappos L, et al. Effects of delayed-release dimethyl fumarate on MRI measures in the phase 3 DEFINE study. J Neurol. 2014;261:1794-1802.
8. O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293-1303.
9. Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13:247-256.
10. Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416-426.
11. Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler. 2018;24:1594-1604.
12. Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387-401.
13. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402-415.
14. Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12:756-767.
15. Kappos L, Li DK, Stüve O, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD Study. JAMA Neurol. 2016;73:1089-1098.
16. Kappos L, Bar-Or A, Cree BA, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomized, phase 3 study. Lancet. 2018;391:1263-1273.
To comment on this article, contact rdavidson@uspharmacist.com.


