US Pharm. 2016;41(6)(Generic Drug Review suppl):6-9.
ABSTRACT: In order to help balance innovation in the pharmaceutical industry with affordable and equitable access to medications, the FDA has typically relied on two key systems: intellectual property (patent) exclusivity and regulatory (market) exclusivity. Although the two systems may seem interchangeable, they are distinct entities rooted in different policies and legislation. As the development of biologic products increases, however, the interplay between these two exclusivities may be at risk. A recent federal appeals court decision has upended the previously well-choreographed exchange between brand and generic manufacturers. Although two federal legislative bills attempt to rectify some of these holes, they fail to address the gap in the patent-dispute process for biologics created by the controversial Amgen v. Sandoz case.
The FDA has long been tasked with balancing innovation in the pharmaceutical industry with affordable and equitable access to medications. To ensure that this balance is maintained, the FDA typically controls the marketing of brand and generic products through two systems: intellectual property exclusivity and regulatory exclusivity. Intellectual property exclusivity takes the form of a patent by the U.S. Patent and Trademark Office, whereas regulatory exclusivity (also known as market exclusivity, data exclusivity, or protection) is at the sole discretion of the FDA. To bring a generic product to market, a manufacturer must either challenge or wait out both of these exclusivities. Today, most disputes around patent and regulatory exclusivities proceed with a high level of transparency around relevant patents, largely because of the Orange Book.
Over the past few decades, legislation and regulations surrounding generic products have transformed how a brand-dominated market translates to a generic-dominated market. Although the Hatch-Waxman Act eased the approval process for generics, there was a necessary shift in focus toward regulatory exclusivity. In the following decades, this shift has created the need for a balancing act—a kind of dance—between generic and brand manufacturers with regard to the two key exclusivities.
In the biologics market, however, the exact interplay between first-to-market (i.e., brand) and follow-on (i.e., generic) has been shaky at best. For instance, the first follow-on biologic product, Zarxio, skipped over the patent-dispute process (also called the patent dance) built into the Biologics Price Competition and Innovation Act (BPCIA).1 In the pursuant legal case, Sandoz argued that the patent-dispute process was not mandated by the plain language. Although the legislation distinctly describes the meticulous steps both parties in a patent dispute should take, the federal appeals court sided with Sandoz and found that the process was not compulsory.2
This article will review high-level details about—and the differences between—patent and regulatory exclusivities, after which the evolution of these exclusivities, as related to the increasing development of biologic products, will be discussed. Finally, some legislative proposals that are pending in Congress (at the time this article was written) will be highlighted.
Differences Between Patent and Regulatory Exclusivities
Pharmaceutical patents may be thought of as the first of two keys necessary in marketing the drug product. In other words, the patent does not give the manufacturer the right to market its product, but instead confers the right to exclude others from making, using, or selling the product. After gaining a patent, in order to receive marketing consent, the manufacturer must show the FDA that the product is safe and effective for use by the public. Once this second key is obtained, the manufacturer can start marketing the product. Although they are relevant to each other, each key, patent, and marketing consent has a distinct set of requirements.
In addition, there are several differences in how patent and regulatory exclusivities are formed, implemented, and enforced. First, patents are not self-enforcing and require action by the patent holder, whereas regulatory exclusivity is enforced by the FDA. In terms of scope, patents are concerned with distinguishing the product’s invention from past disclosures, whereas regulatory exclusivity is centered on the drug product itself. Because patents are drafted and filed ahead of FDA approval, some final products may not even be protected by their patent.3
Patents cannot be obtained if a product is already in the public domain (i.e., is already known and embraced by the public). However, regulatory exclusivity can be conferred onto a manufacturer even if it does not hold a patent for the product. For instance, colchicine has been used to treat gout since ancient times, but in response to the FDA’s unapproved-drugs initiative, Mutual Pharmaceutical Company conducted clinical trials in addition to safety and efficacy trials and received 10 years of regulatory exclusivity. As a result, the price rose from 10 cents to almost $5 per tablet.4
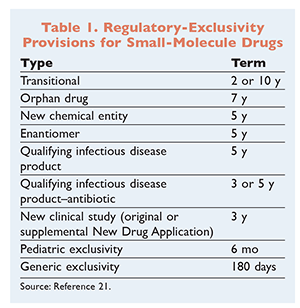
Lastly, regulatory-exclusivity terms are having an increasing effect. After the Hatch-Waxman Act was passed, drug products owed most of their exclusivity to the 20- to 25-year patent-exclusivity term because many regulatory exclusivities were short and did not come close to the 14-year combination restriction (defined in the following section). Based on more recent legislation, however, designations like new chemical entity (NCE), orphan drug, and qualifying infectious disease product can be combined to bring regulatory-exclusivity periods close to the 14-year combination restriction.5-7
About Pharmaceutical Patents
A patent provides an inventor “the grant of a property right ... [for] 20 years from the date on which the application for the patent was filed in the United States.”8 The property right conferred is “the right to exclude others from making, using, offering for sale, or selling the invention in the United States.”8 In other words, the granting of a patent merely confers the right to exclude others from making, using, or selling the product covered by the patent, and the patent is only as powerful as the extent to which the holder can enforce it.8
A pharmaceutical patent is a utility patent, which is granted to manufacturers who invent or discover something “new and useful.”8 A pharmaceutical patent also may be extended by up to 5 years to compensate for any FDA regulatory delays. However, the amount of time that a manufacturer has both patent and regulatory exclusivity cannot exceed 14 years.9 The 14-year combination restriction would be most relevant when an extended regulatory exclusivity is granted (e.g., for an orphan drug).
About Regulatory Exclusivity
The FDA awards regulatory exclusivity to manufacturers of first-to-market brand products and excludes other manufacturers from marketing the drug product for a period of time. The time period differs depending on whether the drug product falls into one or more types of regulatory exclusivity (TABLE 1). For instance, a drug product is an NCE if it contains an active ingredient that has never before been approved by the FDA.5 This effectively delays the time to market for the first generic version of an NCE product, subject to the 14-year combination restriction.
About Biologics
Biologics refers to a class of drug products derived from living organisms, such as “a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product [that is designed for] the prevention, treatment, or cure of a disease or condition of human beings.”10 Although follow-on biologics are alternatives to first-to-market brand products, they are not true generics. In contrast to small-molecule drugs, achieving an exact replication that would yield an interchangeable product is difficult, perhaps even insurmountable, for intellectual property, legal, and chemical reasons. In short, follow-on manufacturers no longer have access to the patents necessary to develop an exact copy of the first-to-market biologic, which usually relies on complex primary, secondary, and tertiary structural nuances to give the product its specific therapeutic and pharmacokinetic properties.11
The BPCIA recognized this challenge by categorizing alternative biologics as either follow-on or interchangeable. An interchangeable designation reflects a higher level of exactness and replication, with “no clinically meaningful differences…in terms of safety, purity, and potency,” whereas a follow-on product is “highly similar” to and follows the same mechanism of action as the reference product.12
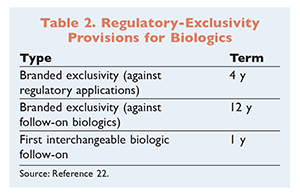
Under the BPCIA, there are three relevant regulatory-exclusivity provisions that apply only to biologics. First, branded products are given a 4-year exclusivity preventing other manufacturers from applying for FDA marketing consensus (TABLE 2). This also prevents other manufacturers from viewing the data contained in the brand-biologic manufacturer’s initial application. Second, branded products are also given a 12-year marketing exclusivity, which effectively prevents the licensing of any follow-on products for 12 years. Lastly, orphan drug exclusivity, which extends drug marketing for 7 years, runs concurrently with the 12-year biologic exclusivity.6 So, if a biologic is also an orphan drug, the exclusivity period is 12 years, not 19 years.
Patent-Dispute Process
Small-Molecule Drugs—the Hatch-Waxman Act: The Hatch-Waxman Act created new rules for patent enforcement and disputes. For instance, under the Bolar exemption, a generic manufacturer can submit an Abbreviated New Drug Application before the expiration of a patent without facing patent-enforcement action by the brand manufacturer.13 In addition, generics manufacturers no longer carry the burden of having to prove safety and efficacy for a drug product, since they can rely on the data submitted by the brand manufacturer.14
The Hatch-Waxman Act also created efficiencies in patent enforcement and disputes for brand manufacturers. For instance, if a brand manufacturer successfully argues a patent-infringement case against a generic manufacturer, the brand manufacturer receives a permanent injunction against the generic manufacturer for the life of its patent.15 Even if a patent-infringement case is brought against a generic manufacturer, the brand manufacturer automatically receives a preliminary injunction against the generic manufacturer for 30 months starting from the initiation of patent litigation.16 This is a significant departure from normal legal practice: A permanent, or even a preliminary, injunction is often difficult to obtain and requires the plaintiff to successfully argue a separate motion.
Biologics—the BPCIA and the Patent Dance: The BPCIA laid out a patent-dispute process—the so-called patent dance because of its many prescribed steps—requiring 1) the follow-on applicant to notify the brand manufacturer of its application; 2) each party to identify relevant patents; 3) an exchange of briefings between parties on the validity of and possible infringement of patents; 4) a negotiation as to which patents will be litigated on; and 5) the simultaneous exchange of patents where negotiations failed.17 This process disallows the involvement of third parties owing to the sensitive nature of materials exchanged and requires strict confidentiality of the representatives involved.17 The process attempts to balance the needs of both sides and level the playing field; however, a major criticism could be that, regardless of confidentiality agreements, manufacturers are handing over sensitive information that gives them a competitive edge.
Amgen v. Sandoz Case: In this case, Sandoz (follow-on manufacturer) forwent this process in order to avoid the exchange and instead to accept a patent-dispute action by Amgen (brand manufacturer). The case went to the federal appeals court, where it was decided by a slim majority that 1) the patent dance was compulsory if the follow-on manufacturer was willing to accept a patent-infringement action immediately and 2) the brand manufacturer was entitled to an automatic 180-day preliminary injunction, preventing the follow-on manufacturer from marketing its product for 180 days.2
In the wake of this decision, follow-on manufacturers have little incentive to participate in the patent dance. The court effectively interpreted the BPCIA as stating that there were two regulatory pathways. Although the patent dance would save some time (e.g., 180-day delay to market), not being required to hand over proprietary information likely is appealing enough to keep some follow-on manufacturers from participating.
Future Developments
Although the federal appeals court held that Sandoz did not have to follow the patent dance, Sandoz filed a writ of certiorari with the Supreme Court to reverse the court’s decision that Sandoz had to wait 180 days after gaining FDA approval before marketing its product. If the Supreme Court decides to hear the case and deems the 180-day waiting period unnecessary, this would boost the entry time of follow-on biologics by up to 180 days.18
Congress has been considering modifications to the regulatory-exclusivity periods. For example, the Modernizing Our Drug and Diagnostics Evaluation and Regulatory Network (MODDERN) Cures Act of 2013 proposed increasing the regulatory-exclusivity term for drugs meeting one or more unmet needs to 15 years.19 Additionally, the 21st Century Cures Act proposes extending regulatory exclusivities by expanding the criteria for orphan drugs and offering additional incentives for antibiotics manufacturers.20 However, the 21st Century Cures Act would be a modest—at best—expansion of regulatory-exclusivity terms.
Alternatively, patent reforms have been proposed through the Innovation Act, but pharmaceutical patents have been excluded from the suggested provisions. These provisions deal with issues that affect electronics, software, and high-technology products but have not occurred with pharmaceutical patents. The fact that none of these proposed reforms applies to pharmaceutical patents may indicate several possible scenarios: 1) The enforcement of pharmaceutical patents may be so tightly linked to regulatory exclusivities that patent reform alone may be insufficient; 2) since competition between drug manufacturers is regulated differently from that in the technology sector, it may be easier to initially address regulatory exclusivities; 3) any real reform may need to address both regulatory exclusivities and patents to create a synergistic effect.
REFERENCES
1. PL 111-148, 124 Stat 119.
2. Amgen v. Sandoz (Fed Cir July 21, 2015) Slip Op at 15.
3. Federal Trade Commission. Generic drug entry prior to patent expiration: an FTC study. www.ftc.gov/sites/default/files/documents/reports/generic-drug-entry-prior-patent-expiration-ftc-study/genericdrugstudy_0.pdf. Accessed May 9, 2016.
4. Rosenthal E. The soaring cost of a simple breath. New York Times. October 12, 2013:A1.
5. 21 USC section 355(j)(5)(F).
6. 21 USC section 360cc.
7. 21 USC section 355f.
8. U.S. Patent and Trademark Office. General information concerning patients. www.uspto.gov/patents-getting-started/general-information-concerning-patents. Accessed March 14, 2016.
9. 35 USC section 156 and section 156(c)3.
10. 42 USC section 262(i).
11. Corbitt AT. The pharmaceutical frontier: extending generic possibilities to biologic therapies in the Biologics Price Competition and Innovation Act of 2007. DePaul J Art Tech Intell Prop Law. 2008;18:365.
12. Wing Yan Tam J. Biologics revolution: the intersection of biotechnology, patent law, and pharmaceutical regulation. Georgetown Law J. 2010;98:535-565.
13. 21 U.S.C. section 271(e)(1).
14. 21 USC section 355(j)(1).
15. 35 USC section 271(e)(4)(A).
16. 21 USC section 355(j)(5)(B)(iii).
17. 42 USC section 262(1)(1)(B)(iii).
18. Sandoz asks Supreme Court to reverse biosimilar decision. www.pharmapatentsblog.com/2016/02/18/sandoz-asks-supreme-court-reverse-biosimilar. Accessed March 14, 2016.
19. HR 3116, MODDERN Cures Act, 113th Congress (2013-2014).
20. HR 6, 21st Century Cures Act, 114th Congress (2015-2016).
21. FDA.gov. Patents and exclusivity. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/UCM447307.pdf. Accessed May 8, 2016.
22. FDA.gov. Biosimilars: additional questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM273001.pdf. Accessed May 8, 2016.
To comment on this article, contact rdavidson@uspharmacist.com.



