US Pharm. 2013;38(10):HS4-HS10.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2012–2013 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction (AR), drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s new drug application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” ARs for many NMEs within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one Black Box Warning for serious ARs or are withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.

Teduglutide (Gattex, NPS Pharmaceuticals)
Indication and Clinical Profile1,2: Teduglutide, an analogue of glucagon-like peptide-2, is approved for the treatment of short bowel syndrome (SBS) in adults requiring parenteral nutrition (PN). Teduglutide is the third drug approved by the FDA to treat SBS in adults receiving nutritional support (Zorbtive [somatropin] and Nutrestore [glutamine] were approved in 2003 and 2004, respectively). In adults, SBS most often results from partial or complete surgical removal of the small and/or large intestine for disorders such as Crohn’s disease. Patients with SBS-associated intestinal failure are unable to absorb sufficient quantities of protein, calories, fluids, electrolytes, and micronutrients, and they often experience diarrhea, weight loss, malnutrition, and dehydration. Accordingly, PN is often necessary in SBS patients; in fact, SBS is the most common diagnosis requiring home PN. Patients with intestinal failure may need PN 5 to 7 days/week for ≥10 hours/day. Unfortunately, PN can lead to serious complications, including hepatobiliary disease, bacteremia, and central venous thromboembolism.
The safety and efficacy of teduglutide were evaluated in two clinical trials and two extension studies that randomized patients to teduglutide treatment or placebo. Clinical response was measured by the number of patients who achieved ≥20% reduction in volume of weekly PN at treatment weeks 20 and 24. In the two clinical trials, 63% and 46% of teduglutide-treated patients achieved clinical response, versus 6% and 30% of placebo patients. After 24 weeks, teduglutide patients had mean PN reductions of 2.5 L/week and 4.4 L/week, compared with 0.9 L/week and 2.3 L/week in placebo patients. The extension studies followed patients who received teduglutide in the initial trials for an additional 28 weeks. These patients experienced mean PN reductions of 4.9 L/week and 5.2 L/week after 1 year of continuous teduglutide treatment, and six patients were weaned off PN while taking teduglutide.
Pharmacology and Pharmacokinetics1,2: Endogenous glucagon-like peptide-2 (GLP-2) is secreted in response to food by intestinal L-cells located primarily in the terminal ileum and the colon. GLP-2 promotes mucosal growth in the small bowel through stimulation of crypt cell proliferation and inhibition of enterocyte apoptosis. It also inhibits gastric acid secretion, reduces gastric motility, enhances mucosal hexose transport, and increases portal and intestinal blood flow. Teduglutide is a 36-membered polypeptide GLP-2 analogue with a longer half-life than that of endogenous GLP-2. It binds to GLP-2 receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial myofibroblasts, and enteric neurons of the submucosal and myenteric plexuses. Activation of these receptors results in local release of multiple mediators, including insulin-like growth factor-1, nitric oxide, and keratinocyte growth factor, resulting in increased villus height and crypt depth of the intestinal mucosa and enhanced gastrointestinal (GI) fluid absorption.
Subcutaneous (SC) teduglutide has an absolute bioavailability of 88% and provides maximum plasma concentrations at 3 to 5 hours after administration. Although the metabolism of teduglutide has not been fully elucidated, the drug appears to be degraded into small peptides and amino acids, similarly to the catabolism of endogenous GLP-2. Teduglutide’s plasma clearance, which is approximately 123 mL/h/kg, is similar to the glomerular filtration rate, suggesting that teduglutide is cleared primarily renally. Teduglutide has a mean terminal half-life of approximately 2 hours in healthy patients and 1.3 hours in SBS patients. In patients with moderate-to-severe renal impairment or end-stage renal disease (ESRD), the maximum concentration of drug and AUC0-inf of teduglutide are significantly increased (>2-fold), suggesting the need for dosage reductions in these patients.
Adverse Reactions (ARs) and Drug Interactions1,2: The most common ARs in clinical trials were abdominal pain, injection-site reactions, nausea, headaches, stoma complication, and abdominal distention. Antiteduglutide antibodies developed in a few patients, but did not appear to reduce the drug’s effect. Other ARs included intestinal obstruction, biliary-tract disorders, and increased incidence of upper respiratory tract infection. Because teduglutide may accelerate the growth of GI cancers, colonoscopy should be performed before teduglutide initiation and repeated after 1 year, with subsequent examinations as needed but no less frequently than every 5 years. To ensure that the benefits of teduglutide outweigh the potential risks, the drug was approved with a Risk Evaluation and Mitigation Strategy consisting of a communication plan and training for prescribers. Furthermore, the FDA is requiring a postmarketing study of SBS patients receiving teduglutide in a routine clinical setting to further evaluate the drug’s potential to increase the risk of colorectal cancer and other conditions. Teduglutide is classified as Pregnancy Category B.
Based on teduglutide’s mechanism, there is a potential for increased absorption of concomitant oral medications that should be considered if these drugs require titration or have a narrow therapeutic index (e.g., benzodiazepines, phenothiazines). No inhibition or induction of CYP450 has been observed in in vitro studies.
Dosage and Administration1,2: Teduglutide is supplied in single-use glass vials containing 5 mg of the drug as a lyophilized powder. Upon reconstitution with the 0.5 mL Sterile Water for Injection provided in the prefilled syringe, the solution delivers a maximum of 3.8 mg of teduglutide. The recommended daily dosage is 0.05 mg/kg body weight administered by SC injection once daily. A 50% dosage reduction is recommended in patients with moderate-to-severe renal impairment (creatinine clearance <50 mL/min) or ESRD. It is recommended to alternate sites for SC injection. Possible sites include the thighs, arms, and abdominal quadrants. Teduglutide should not be administered IV or intramuscularly. If a dose is missed, it should be administered as soon as possible that day, but two doses should not be administered on the same day.
Colonoscopy (or alternative imaging) of the entire colon, with removal of polyps, should be performed within 6 months prior to teduglutide initiation, and follow-up colonoscopy is recommended as described above. Patients should also undergo initial laboratory assessments (bilirubin, alkaline phosphatase, lipase, and amylase) within 6 months prior to treatment initiation and every 6 months thereafter. If clinically meaningful elevation of these values occurs, further imaging of the biliary tract, liver, or pancreas may be necessary. Discontinuation of teduglutide may result in fluid and electrolyte imbalance; therefore, patients’ fluid and electrolyte status should be carefully monitored.
Ponatinib (Iclusig, Ariad Pharmaceuticals)
Indication and Clinical Profile3,4: Ponatinib is a new multitargeted tyrosine kinase inhibitor (TKI) approved as an orphan drug under the FDA’s accelerated approval program. It is indicated to treat patients with chronic-, accelerated-, or blast-phase chronic myeloid leukemia (CML) or Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ALL) that is resistant or intolerant to prior TKI therapy. CML and Ph+ALL are WBC cancers that affect more than 20,000 people in the United States. They are typically asymptomatic initially, but an enlarged spleen, fatigue, paleness from anemia, and disrupted thermoregulation may develop. Ponatinib is the third drug approved for CML treatment since 2012; the others are bosutinib (Bosulif) and omacetaxine (Synribo).
Approval of ponatinib was based on a single-arm, open-label, multicenter, phase II trial involving >449 patients with CML or Ph+ALL whose disease was resistant or intolerant to prior TKI therapy. Ponatinib 45 mg once daily resulted in a reduction in the percentage of cells expressing the Philadelphia chromosome genetic mutation (major cytogenetic response) in 54% of patients, including 70% of patients who also had a T315I mutation. Ponatinib also resulted in a normalization of WBC counts or no evidence of leukemia (major hematologic response) in 52% of accelerated-phase CML patients, 31% of blast-phase CML patients, and 41% of Ph+ALL patients.
Pharmacology and Pharmacokinetics3,4: Ponatinib is an imidazo[1,2-b]pyridazine benzamide (FIGURE 1) that functions as a TKI. Its primary target is the breakpoint cluster region–Abelson (Bcr-Abl) tyrosine kinase expressed by CML and Ph+ALL, but it also targets Bcr-Abl isoforms that carry mutations, including the T315I mutation. Ponatinib also binds to members of the VEGFR, PDGFR, FGFR, and EPH receptors and SRC families of kinases, and to KIT, RET, TIE2, and FLT3.
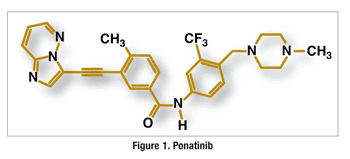
The absolute bioavailability of ponatinib is unknown. The drug has a half-life of 24 hours, and it is >99% plasma protein bound. Ponatinib is metabolized by CYP3A4 and, to a lesser extent, by CYP2C8, 2D6, 3A5, and esterases and amidases. Approximately 64% of ponatinib is excreted as metabolites, primarily in the feces. Ponatinib does not inhibit metabolism of other CYP-substrate drugs, nor does it appear to induce CYP metabolism. It does, however, inhibit adenosine triphosphate–binding cassette (ABC) G2, the bile salt export pump, and the P-glycoprotein (Pgp) pump.
Adverse Reactions (ARs) and Drug Interactions3,4: The most common ARs (≥20%) reported in clinical trials were myelosuppression (thrombocytopenia, neutropenia, leukopenia, anemia, and lymphopenia), hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, arthralgia, nausea, and pyrexia. Ponatinib also carries a Black Box Warning that the drug may cause arterial thrombosis and hepatotoxicity. The drug should be avoided in pregnancy because of its potential for fetal harm (Pregnancy Category D). Patients who breastfeed or intend to breastfeed should not use ponatinib.
The ponatinib dosage should be reduced when the drug is coadministered with CYP3A inhibitors. Coadministration of ponatinib with strong CYP3A inducers or drugs that elevate gastric pH (proton pump inhibitors, H2 blockers, antacids) may lead to inadequate drug concentration. Because of its inhibition of Pgp and ABC, ponatinib may result in altered efficacy of drugs that are sensitive substrates of these pumps; however, formal studies have not been conducted to determine the severity of this interaction.
Dosage and Administration3,4: Ponatinib is supplied as 15-mg and 45-mg tablets. The recommended dosage is 45 mg once daily with or without food. The dosage should be reduced to 30 mg once daily when the drug is administered with strong CYP3A inhibitors. Ponatinib has not been studied in pediatric patients or in hepatically or renally impaired patients. Ponatinib therapy should be interrupted for 1 week prior to major surgery to avoid an increased bleeding risk. Patients taking ponatinib who experience myelosuppression, hepatic toxicity, asymptomatic lipase elevation (grade 3-4), or pancreatitis (grade 2-3) should interrupt therapy until toxicity subsides, then resume therapy at the dosage recommended by the manufacturer.
Omacetaxine (Synribo, Teva Pharmaceuticals USA)
Indication and Clinical Profile5,6: Omacetaxine mepesuccinate is a protein synthesis inhibitor approved as an orphan drug under the FDA’s accelerated approval program. It is indicated for the treatment of chronic myelogenous leukemia (CML) (see discussion in Ponatinib section) that is resistant or intolerant to two or more tyrosine kinase inhibitors (TKIs). Omacetaxine is one of three drugs approved for the treatment of CML since 2012; the others are bosutinib (Bosulif) and ponatinib (Iclusig).
Approval of omacetaxine was based on research involving a combined cohort of patients whose cancer progressed after previous treatment with two or more TKIs. Omacetaxine’s effectiveness against chronic-phase CML was demonstrated by a reduction in the percentage of cells expressing the Philadelphia chromosome genetic mutation found in most CML patients. Fourteen of 76 patients (18.4%) achieved reduction in an average of 3.5 months. Median length of reduction was 12.5 months. In accelerated-phase CML, omacetaxine’s effectiveness was determined by the number of patients who experienced WBC-count normalization or had no evidence of leukemia (major hematologic response [MaHR]). Five of 35 patients (14.3%) achieved MaHR in an average of 2.3 months. Median MaHR duration in these patients was 4.7 months.
Pharmacology and Pharmacokinetics5,6: Omacetaxine is a cephalotaxine ester (FIGURE 2) prepared by a semisynthetic process from the leaves of Cephalotaxus harringtonia. It functions as a protein translation inhibitor by preventing the initial elongation step of protein synthesis through binding to the large ribosomal subunit A-site cleft. This prevents correct positioning of amino acid side chains of aminoacyl-tRNAs. In vitro, omacetaxine has been shown to reduce protein levels of the Bcr-Abl oncoprotein and the Mcl-1 antiapoptotic protein. Because it acts at the initial stage of protein synthesis, however, omacetaxine has no effect on protein synthesis from mRNAs already undergoing translation.
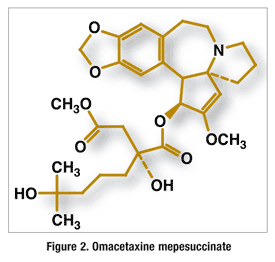
The absolute bioavailability of omacetaxine is unknown. The drug has a steady-state volume of distribution of about 140 L and a half-life of 6 hours, and it is ≤50% plasma protein bound. Omacetaxine’s primary route of metabolism is hydrolysis to 4´-DMHHT via plasma esterases, with little hepatic microsomal oxidative or esterase-mediated metabolism. The major route of elimination is currently unknown; however, an average of 15% is excreted unchanged in the urine. Omacetaxine does not inhibit metabolism of CYP450 substrates, and its ability to induce CYP450 enzymes has not been determined.
Adverse Reactions (ARs) and Drug Interactions5,6: The most common ARs (≥20%) reported in clinical trials were myelosuppression (thrombocytopenia, anemia, neutropenia, lymphopenia), diarrhea, nausea, fatigue, pyrexia, asthenia, infection, and injection-site reactions. The package insert includes a warning about omacetaxine’s ability to induce hyperglycemia, although less common (11%), and a precaution about its use in diabetic patients without good glycemic control. Omacetaxine should be avoided in pregnant women because of its potential for fetal harm (Pregnancy Category D). Also, women who breastfeed or intend to breastfeed should not use omacetaxine.
In vitro drug-interaction studies with omacetaxine revealed no significant interactions. However, concomitant use of anticoagulants, aspirin, or nonsteroidal anti-inflammatory drugs (NSAIDs) may increase the risk of bleeding.
Dosage and Administration5,6: Omacetaxine is supplied in 3.5-mg single-use vials as a lyophilized powder to be dissolved into solution for subcutaneous administration. The induction dosage of omacetaxine is 1.25 mg/m2 twice daily for 14 consecutive days over a 28-day cycle, followed by a maintenance dosage of 1.25 mg/m2 twice daily for 7 consecutive days over a 28-day cycle, which should be repeated as long as the patient continues to benefit from therapy. Because of insufficient clinical data, there is no recommended dosage adjustment for renally or hepatically impaired patients or for pediatric patients. There is no recommended dosage adjustment for elderly patients.
Cabozantinib (Cometriq, Exelixis)
Indication and Clinical Profile7,8: Cabozantinib is a new tyrosine kinase inhibitor (TKI) approved to treat metastatic medullary thyroid cancer (MTC). The National Cancer Institute estimated that, in 2012, 56,460 people in the United States would be diagnosed with thyroid cancer and nearly 2,000 would die from it. Only about 4% of thyroid cancers are MTC. MTC develops in thyroid parafollicular cells, which produce calcitonin (which regulates plasma calcium levels). MTC appears to occur spontaneously or in families with certain genetic mutations that lead to one or more cancers of the endocrine system. Cabozantinib is the second drug approved to treat MTC in the past 2 years; vandetanib (Caprelsa) was approved in April 2011. Cabozantinib was approved in 6 months under the FDA’s priority review program, which allows an expedited review for drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists. Cabozantinib also was designated an orphan product because it was developed to treat a rare disease.
The safety and effectiveness of cabozantinib were established in an international randomized, controlled trial involving 330 subjects with MTC. Subjects were required to have evidence of actively progressive disease within 14 months prior to study entry that was confirmed by an Independent Radiology Review Committee or the treating physician. Subjects received cabozantinib 140 mg or placebo orally once daily without food until disease progression or intolerable toxicity. Randomization was stratified by age (<65 years vs. >65 years) and prior TKI use. The main efficacy outcome measures were progression-free survival (PFS), objective response (OR), and response duration. The cabozantinib arm had a statistically significant prolongation of PFS compared with the placebo arm (median PFS of 11.2 months and 4.0 months, respectively). Partial responses were observed only in the cabozantinib arm (27% vs. 0%). Median duration of OR was 14.7 months for subjects treated with cabozantinib, and there was no statistically significant difference in overall survival between the treatment arms at the planned interim analysis.
Pharmacology and Pharmacokinetics7,8: Cabozantinib is a small-molecule inhibitor (FIGURE 3) of tyrosine kinases (TKs), including RET; MET; VEGFR-1, -2, and -3; KIT; TRKB; FLT-3; AXL; and TIE-2. These receptor TKs are involved in both normal cellular function and pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment. Treatment with cabozantinib has been shown to reduce tumor growth, metastasis, and angiogenesis.
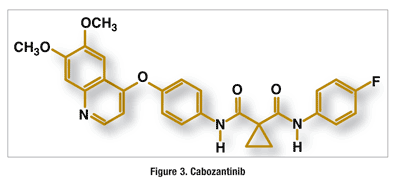
Following oral administration, median time to peak cabozantinib plasma concentrations (Tmax) ranges from 2 to 5 hours. The drug is metabolized by CYP3A4 and is eliminated primarily in the feces. Because of the narrow therapeutic window and this clearance profile, CYP3A4 inducers and inhibitors should be avoided. The predicted effective half-life is approximately 55 hours, oral volume of distribution is approximately 349 L, and clearance at steady state is estimated to be 4.4 L/h.
Adverse Reactions (ARs) and Drug Interactions7,8: The most common ARs reported with cabozantinib include diarrhea; mouth sores; redness, pain, or swelling of the digits; loss of appetite and weight loss; nausea; fatigue; graying or loss of hair color; taste alteration; hypertension; abdominal pain; and constipation. The package insert for cabozantinib includes a Black Box Warning concerning severe and fatal bleeding and perforations and fistula in the colon, which occurred in some trial patients. The most common laboratory abnormalities are increased liver enzymes and decreased calcium, phosphorus, WBCs, and platelets. Cabozantinib can cause fetal harm and thus is Pregnancy Category D. A decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Based on the clearance profile, strong CYP3A4 inhibitors (azole antifungals, protease inhibitors, clarithromycin, telithromycin) should be avoided with cabozantinib. Chronic coadministration of strong CYP3A4 inducers (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital, St. John’s wort) should also be avoided. Cabozantinib is an inhibitor, but not a substrate, of P-glycoprotein (Pgp) transport; thus, it has the potential to increase plasma concentrations of coadministered Pgp substrates.
Dosage and Administration7,8: Cabozantinib is supplied as 20-mg and 80-mg capsules. The recommended daily dosage is 140 mg (one 80-mg and three 20-mg capsules). Capsules should be swallowed whole. Patients should not eat ≥2 hours before and ≥1 hour after taking cabozantinib. Treatment should be continued until disease progression or unacceptable toxicity occurs. Foods (e.g., grapefruit products), nutritional supplements, and drugs known to inhibit or induce CYP450 should be avoided. Cabozantinib use is not recommended in moderate-to-severe hepatic impairment.
Bedaquiline (Sirturo, Janssen Therapeutics)
Indication and Clinical Profile9,10: Bedaquiline was approved as part of combination therapy for adults with multidrug-resistant pulmonary tuberculosis (MDR-TB) when other alternatives are not available. TB, which is caused by Mycobacterium tuberculosis, is one of the world’s deadliest diseases. The first drug approved to treat MDR-TB, bedaquiline should be used in combination with other antituberculars. The FDA granted bedaquiline fast-track, priority-review, and orphan-product status because the drug could fill an unmet medical need, could potentially provide safe and effective treatment where no satisfactory alternative therapy exists, and was developed to treat a rare disease. Bedaquiline is not indicated to treat latent, extrapulmonary, or drug-sensitive TB.
Bedaquiline’s safety and effectiveness were established in 440 patients in two phase II clinical trials. Patients in the first trial were randomized either to bedaquiline plus other drugs used to treat TB or to placebo plus other drugs used to treat TB. All patients in the second trial, which is ongoing, received bedaquiline plus other TB drugs. Both trials measured the length of time it took for a patient’s sputum to be free of M tuberculosis (sputum culture conversion [SCC]). In the first trial, patients receiving bedaquiline combination therapy achieved SCC in a median of 83 days, compared with 125 days in patients receiving placebo combination therapy. In the second trial, the median time to SCC was 57 days, supporting the efficacy findings of the first trial. There are no clinical data on the combined use of antiretroviral agents and bedaquiline in HIV/MDR-TB–coinfected patients.
Pharmacology and Pharmacokinetics9,10: Bedaquiline is a diarylquinoline (FIGURE 4) that inhibits mycobacterial adenosine 5´-triphosphate synthase, an enzyme that regulates the proton pump of adenosine triphosphate synthase and is essential for the generation of energy and mycobacterial replication.
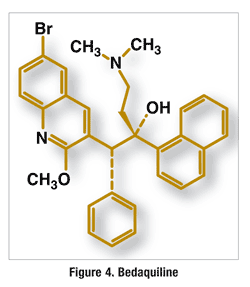
Bedaquiline is slowly absorbed from the gastrointestinal tract and should be taken with food to enhance its oral bioavailability. It is highly bound by plasma proteins and is widely distributed (volume of distribution 164 L). CYP3A4 is the major isoenzyme involved in the metabolism of bedaquiline, and the primary metabolite formed, N-monodesmethyl-bedaquiline, has significantly reduced antimycobacterial activity. Bedaquiline and its metabolites are eliminated primarily in feces. Based on this clearance profile, bedaquiline should be used with caution in patients with severe hepatic impairment and only when the benefits outweigh the risks. Also, since bedaquiline is highly bound to plasma proteins, it is unlikely that it will be significantly removed from plasma by hemodialysis (HD) or peritoneal dialysis (PD). Therefore, it should be used with caution in patients with severe renal impairment or end-stage renal disease requiring HD or PD.
Adverse Reactions (ARs) and Drug Interactions9,10: The most common ARs in clinical studies were nausea, joint and chest pain, and headache. Some liver-related ARs were also reported in clinical trials; thus, alcohol and other hepatotoxic drugs should be avoided, especially in patients with diminished hepatic reserve. Bedaquiline has a Black Box Warning about its potential to cause fatal arrhythmias, since it may induce long QT syndrome by blocking the hERG channel. The manufacturer is distributing the drug from a single source and is providing educational materials to help ensure that it is used appropriately. Bedaquiline is Pregnancy Category B, and animal studies have shown it to be concentrated in breast milk. Because of the potential for ARs in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug.
Because bedaquiline is metabolized by CYP3A4, its systemic exposure and therapeutic efficacy may be reduced if it is coadministered with CYP3A4 inducers, including rifamycins (e.g., rifampin, rifapentine, rifabutin). Therefore, strong CYP3A4 inducers should be avoided in patients taking bedaquiline. Also, coadministration of bedaquiline with strong CYP3A4 inhibitors may increase systemic exposure to bedaquiline, which can potentially increase the risk of ARs. Therefore, the use of strong CYP3A4 inhibitors for >14 consecutive days should be avoided with bedaquiline, unless the benefit of treatment with the drug combination outweighs the risk.
Dosage and Administration9,10: Bedaquiline is supplied as a 100-mg tablet. It should be used only in combination with at least three other drugs to which the patient’s MDR-TB isolate has been shown to be susceptible in vitro. If in vitro testing results are unavailable, treatment may be initiated with bedaquiline in combination with at least four other drugs to which the patient’s MDR-TB isolate is likely to be susceptible. The recommended dosage for the first 2 weeks is 400 mg once daily with food. This is followed, in weeks 3 to 24, with a dosage of 200 mg three times per week with food, for a total of 600 mg per week.
REFERENCES
1. Gattex (teduglutide [rDNA origin]) product information. Bedminster, NJ: NPS Pharmaceuticals; December 2012.
2. Jeppesen PB, Pertkiewicz M, Messing B, et al. Teduglutide reduces
need for parenteral support among patients with short bowel syndrome
with intestinal failure. Gastroenterology. 2012;143:1473-1481.
3. Iclusig (ponatinib) product information. Cambridge, MA: Ariad Pharmaceuticals, Inc; December 2012.
4. Zhou T, Commodore L, Huang WS, et al. Structural mechanism of the
Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase
inhibitor resistance. Chem Biol Drug Des. 2011;77:1-11.
5. Synribo (omacetaxine mepesuccinate) product information. North Wales, PA: Teva Pharmaceuticals USA, Inc; October 2012.
6. Cortes J, Lipton JH, Rea D; Omacetaxine 202 Study Group. Phase 2
study of subcutaneous omacetaxine mepesuccinate after TKI failure in
patients with chronic-phase CML with T315I mutation. Blood. 2012;120:2573-2580.
7. Cometriq (cabozantinib) product information. San Francisco, CA: Exelixis, Inc; November 2012.
8. Sherman SI, Cohen EE, Schöffski P, et al. Efficacy of cabozantinib
(Cabo) in medullary thyroid cancer (MTC) in patients with RAS or RET mutations: results from a phase III study. J Clin Oncol. 2013;31(suppl) [abstract 6000].
9. Sirturo (bedaquiline) product information. Titusville, NJ: Janssen Therapeutics; December 2012.
10. Diacon AH, Donald PR, Pym A, et al. Randomized pilot trial of
eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant
tuberculosis: long-term outcome, tolerability, and effect on emergence
of drug resistance. Antimicrob Agents Chemother. 2012;56:3271-3276.
To comment on this article, contact rdavidson@uspharmacist.com.



