US Pharm. 2020;45(10):HS-1-HS-8.
ABSTRACT: New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2020 (TABLE 1) detail the basic clinical and pharmacologic profile for each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, often “new” adverse reactions for many NMEs emerge within 2 to 3 years of their first becoming available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Ozanimod (Zeposia, Bristol-Myers Squibb)
Indication and Clinical Profile1,2: Ozanimod is approved for the treatment of adults with relapsing forms of multiple sclerosis (RMS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease. Multiple sclerosis (MS), a disease affecting nearly one million people in the United States, results from the immune system attacking the protective myelin sheath of nerves, creating lesions that interrupt the signaling between nerves, causing the symptoms and relapses. Treatment with disease-modifying therapies can reduce the number of disease attacks, but typically MS patients respond differently to these medications. Therefore, it is important to have a variety of treatment options. Ozanimod, a once-daily oral medication, is the only approved sphingosine-1-phosphate (S1P) receptor modulator that offers RMS patients an initiation with no genetic test and no label-based, first-dose observation requirement.
The FDA approval of ozanimod is based on two randomized, controlled phase III trials labeled SUNBEAM and RADIANCE. SUNBEAM evaluated the efficacy and tolerability of oral ozanimod (Zeposia)(0.92 mg, equivalent to 1 mg) against weekly IM interferon beta-1a (Avonex) for at least a 12-month treatment period. The study included 1,346 people with RMS across 152 sites in 20 countries. The primary endpoint of the trial was annualized relapse rates (ARR) during the treatment period. RADIANCE Part B evaluated the efficacy, safety, and tolerability of ozanimod over the same dose range versus weekly IM interferon beta-1a over a 24-month treatment period. This study included 1,320 people with RMS across 150 sites in 21 countries with a primary endpoint of ARR over 24 months. Ozanimod demonstrated a relative reduction in ARR versus interferon beta-1a of 48% through 1 year and 38% at 2 years. At one year, treatment with ozanimod reduced the number of T1-weighted gadolinium-enhanced (GdE) brain lesions more than interferon beta-1a (0.16 vs. 0.43), a relative reduction of 63%, and reduced the number of new or enlarging T2 lesions (1.47 vs. 2.84), a relative reduction of 48%. At 2 years, treatment reduced the number of T1-weighted GdE brain lesions more than interferon beta-1a (0.18 vs. 0.37), a relative reduction of 53%. Treatment also reduced the number of new or enlarging T2 lesions versus interferon beta-1a (1.84 vs. 3.18) with a relative reduction of 42%. There was no statistically significant difference in the 3-month and 6-month confirmed disability progression between ozanimod-treated and interferon beta-1a–treated patients over 2 years.
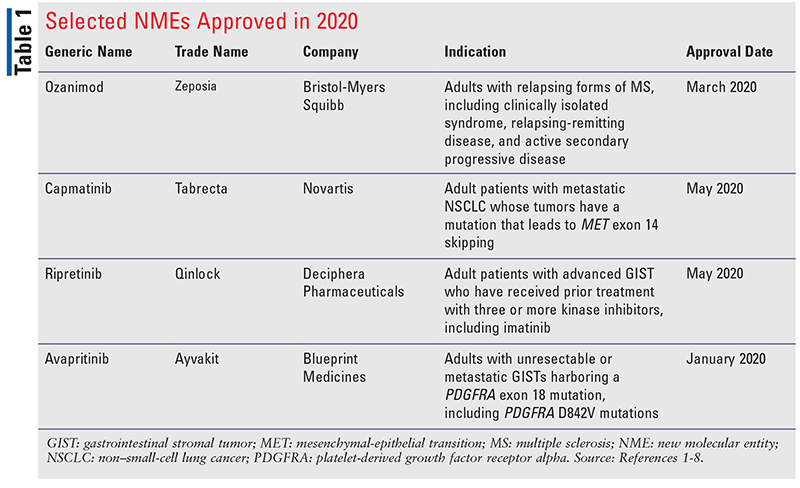
Pharmacology and Pharmacokinetics1,2: The mechanism by which ozanimod exerts therapeutic effects in MS has not been fully established but appears to involve the reduction of lymphocyte migration into the central nervous system. Ozanimod is an indenyl-oxadiazole (FIGURE 1) designed to function as a sphingosine 1-phosphate (S1P) receptor modulator, binding with high affinity to S1P receptor subtypes 1 and 5. In doing so, the drug inhibits the ability of lymphocytes to migrate from lymph nodes, and thereby reduces the number of lymphocytes in the circulation.
Ozanimod has high oral bioavailability, reaching peak plasma concentrations at 6 to 8 hours. No clinically significant differences in the Cmax and area under the curve (AUC) of ozanimod were observed following administration with a high-fat, high-calorie meal. Ozanimod is metabolized by multiple enzymes, including CYP3A4 and CYP2C8, and various dehydrogenases to form circulating major active metabolites (e.g., CC112273 and CC1084037) and several minor active metabolites with similar selectivity for the target receptors. Approximately 94% of circulating total active drug/metabolite includes ozanimod (6%) and its metabolites CC112273 (73%), and CC1084037 (15%). The mean apparent volume of distribution is 5590 L, and plasma protein binding of the parent drug and its metabolites CC112273 and CC1084037 is greater than approximately 98%. The drug and its metabolites are excreted in both the urine and the feces. The mean plasma half-life of ozanimod is approximately 21 hours, while its active metabolites have half-lives exceeding 10 days.
Adverse Reactions and Drug Interactions1-2: The most common adverse reactions (³4%) associated with ozanimod in clinical trials included upper respiratory infection, hepatic transaminase elevation, orthostatic hypotension, urinary tract infection, back pain, and hypertension. The drug’s label also includes Warnings and Precautions for increased risk of infections, bradyarrhythmia and atrioventricular conduction delays, liver injury, fetal risk, increased blood pressure, respiratory effects, macular edema, posterior reversible encephalopathy syndrome, and additive immunosuppressive effects from prior immune-modulating treatments. There is also a warning for severe increase in disability and immune system effects after stopping ozanimod. Based on these precautions, prior to initiation of ozanimod therapy, all patients should have a recent complete blood count including lymphocyte count (within 6 months or after discontinuation of prior MS therapy), an ECG to identify preexisting conduction abnormalities, a recent liver function test (within 6 months), and consideration of current and prior medications, including vaccinations. For patients with a history of uveitis or macular edema, an ophthalmic assessment is required. Ozanimod is contraindicated in patients who in the last 6 months experienced myocardial infarction, unstable angina, stroke, transient ischemic attack, decompensated heart failure requiring hospitalization, or class III/IV heart failure; patients who have a presence of Mobitz type II second-or third-degree atrioventricular block, sick sinus syndrome, or sinoatrial block (unless the patient has a functioning pacemaker); those with severe untreated sleep apnea; and patients taking a monoamine oxidase inhibitor.
Caution is advised in pregnancy since administration in animals during pregnancy produced adverse effects on development (including embryolethality, an increase in fetal malformations, and neurobehavioral changes) in the absence of maternal toxicity. Because of the time required to eliminate the drug from the body, the potential risk to the fetus may persist, and women of childbearing age should use effective contraception for 3 months after stopping ozanimod. Ozanimod and/or its metabolites have been detected in the milk of lactating rodents. Thus the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ozanimod and any potential adverse effects on the breastfed infant.
The use of live attenuated vaccines should be avoided during and for up to 3 months after treatment with ozanimod. The coadministration of strong CYP2C8 inhibitors and breast cancer resistance protein (BCRP) inhibitors is not recommended, and concurrent use of strong CYP2C8 inducers should be avoided. Coadministration of ozanimod with monoamine oxidase B (MAO-B )inhibitors may decrease exposure of the active metabolites of ozanimod. In addition, metabolites of ozanimod may inhibit MAO, resulting in hypertensive crises. Therefore, coadministration with MAO inhibitors (e.g., selegiline, phenelzine, linezolid) is contraindicated, and at least 14 days should elapse between discontinuation of ozanimod and initiation of treatment with MAO inhibitors. Also, coadministration of ozanimod with drugs or OTC medications that can increase norepinephrine or serotonin (opioids, selective serotonin reuptake inhibitors, selective norepinephrine reuptake inhibitors, tricyclics, tyramine) is not recommended, and patients using such drugs should be monitored concurrently for hypertension.
Dosage and Administration1,2: Ozanimod is supplied as 0.23-mg, 0.46-mg, and 0.92-mg capsules for oral administration and dosed based on the Warnings and Precautions listed above. Therapy typically is initiated with a 7-day titration where 0.23 mg once daily is administered on days 1–4 and 0.46 mg once daily on days 5–7. On day 8 and thereafter, a maintenance dose of 0.92 mg once daily is recommended. Ozanimod capsules should be swallowed whole and can be administered with or without food. If a dose is missed during the first 2 weeks of treatment, treatment may be reinitiated using the titration regimen. If a dose is missed after the first 2 weeks, treatment should continue as planned.
Capmatinib (Tabrecta, Novartis)
Indication and Clinical Profile3,4: Capmatinib is specifically indicated for the treatment of adult patients with metastatic non–small-cell lung cancer (NSCLC) whose tumors have a mutation that leads to mesenchymal-epithelial transition (MET) exon 14 skipping. This drug was approved along with a FoundationOne CDx assay (F1CDx) as a companion diagnostic to detect the mutation. NSCLC is the most common type of lung cancer, with up to 90% of all lung carcinomas falling into the non–small-cell category. Mutations leading to MET exon 14 skipping are found in 3% to 4% of patients with lung cancer. With this form of cancer, there is a high likelihood that the cancer cells will metastasize from the lungs to other organs, and MET exon 14 skipping is recognized as a critical event for metastasis of carcinomas. Capmatinib was approved under the Accelerated Approval pathway, which provides for the approval of drugs that treat serious or life-threatening diseases and generally provide a meaningful advantage over existing treatments. The FDA also granted Breakthrough Therapy and Orphan Drug designation for this drug.
The FDA approval of capmatinib was based on results from the pivotal GEOMETRY mono-1 phase II multicenter, multicohort study in adult patients with EGFR wild-type, metastatic NSCLC as measured by overall response rate (ORR). The trial evaluated 97 adult patients with metastatic NSCLC harboring confirmed mutations that lead to METex14, who were assigned to cohorts 4 (n = 69, previously treated patients) or 5b (n = 28, treatment-naive) and who received capmatinib tablets 400 mg orally twice daily. In the METex14 population (n = 97), the confirmed ORR was 68% and 41% among treatment-naive (n = 28) and previously (n = 69) treated patients, respectively, based on the Blinded Independent Review Committee assessment per RECIST v1.1. In patients taking capmatinib, the study also demonstrated a median duration of response of 12.6 months in treatment-naive patients (19 responders) and 9.7 months in previously treated patients (28 responders). Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
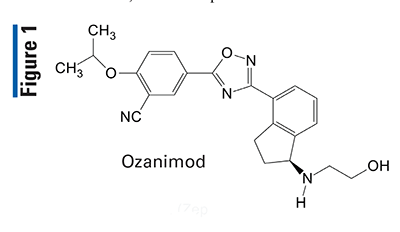
Pharmacology and Pharmacokinetics3,4: Capmatinib is quinolone-imidazo-benzamide (FIGURE 2) designed as a MET kinase inhibitor, including the mutant variant produced by exon 14 skipping. MET exon 14 skipping results in a protein with a missing regulatory domain that reduces its negative regulation leading to increased downstream MET signaling and cell proliferation.
The absorption of capmatinib after oral administration is greater than 70%, giving peak plasma concentrations (Cmax) in approximately 1-2 hours (Tmax). High-fat meals reportedly increase the area under the curve (AUC) by 46% with no change in Cmax. Capmatinib reaches steady state by day 3 following twice-daily dosing. Plasma protein binding is 96%, and the apparent mean volume of distribution at steady state is 164 L. Capmatinib is primarily metabolized by CYP3A4 and aldehyde oxidase. The drug and its metabolites are primarily excreted in the feces with an effective elimination half-life of 6.5 hours.
Adverse Reactions and Drug Interactions3,4: Common adverse effects observed in at least 20% of trial patients included peripheral edema, nausea, fatigue, vomiting, shortness of breath, and decreased appetite. Capmatinib may cause serious side effects, including interstitial lung disease or pneumonitis, requiring permanent drug discontinuation. The drug may also cause hepatotoxicity, and therefore liver function tests should be obtained prior to starting therapy. If a patient experiences hepatotoxicity, capmatinib should be withheld, the dose reduced, or the drug permanently discontinued. Based on a clear positive signal for phototoxicity in laboratory studies, patients may be more sensitive to sunlight and should be advised to take precautions to cover their skin and use sunscreen and not to tan while taking capmatinib. Pregnant women should be advised that capmatinib may cause harm to a developing fetus or newborn baby. Also, both females of reproductive potential and male patients with female partners of reproductive potential should be counseled to use effective contraception during treatment with this drug and for 1 week after the last dose.
Dosage and Administration3,4: Capmatinib is supplied as 150-mg and 200-mg tablets for oral administration. The recommended dosage is 400 mg orally twice daily with or without food. The tablets should be swallowed whole and not broken, crushed, or chewed. If a dose is missed or vomiting occurs after administration, the patient should not make up the dose but take the next dose at its scheduled time. To manage adverse reactions, a dose reduction to 300 mg twice daily or 200 mg twice daily is recommended for the first and second incidence of adverse effects, respectively. Capmatinib should be discontinued for more serious adverse reactions, such as interstitial lung disease and grade 4 indicators of hepatic injury.
Ripretinib (Qinlock, Deciphera Pharmaceuticals)
Indication and Clinical Profile5,6: Ripretinib is specifically indicated for the treatment of adult patients with advanced gastrointestinal stromal tumor (GIST) who have received prior treatment with three or more kinase inhibitors, including imatinib. GISTs arise when abnormal cells form in the tissues of the gastrointestinal tract. Each year 4,000 to 6,000 adults in the United States. are diagnosed with a GIST. Over the past 20 years, four FDA-approved kinase inhibitor therapies targeting GIST have been approved: imatinib in 2002, sunitinib in 2006, regorafenib in 2013, and avapritinib in 2020. However, some patients do not respond to these treatments, and their tumors continue to progress. The approval of ripretinib provides a new treatment option for patients who have exhausted all FDA-approved therapies for GIST.
FDA approval of ripretinib was based on INVICTUS, a phase III randomized, placebo-controlled, international study evaluating the tolerability and efficacy of this drug compared with placebo in patients with advanced GIST whose previous therapies have included imatinib, sunitinib, and regorafenib. Patients were randomized 2:1 to either 150 mg of ripretinib or placebo once daily in 28-day cycles and repeated until tumor growth was found (disease progression) or the patient experienced intolerable side effects. The primary efficacy endpoint was progression-free survival (PFS) as determined by independent radiologic review using modified Response Evaluation Criteria in Solid Tumors (RECIST). The median PFS in the study was 6.3 months compared with 1 month in the placebo arm and significantly reduced the risk of disease progression or death by 85%. Secondary endpoints, as determined by independent radiologic review using modified RECIST, included objective response rate (ORR) and overall survival (OS). Ripretinib demonstrated an ORR of 9.4% compared with 0% for placebo and a median OS of 15.1 months compared with 6.6 months in the placebo arm, with risk of death reduced by 64%. After disease progression, patients who were randomized to placebo were given the option of switching to ripretinib. The FDA granted this application Priority Review and Fast Track, as well as Breakthrough Therapy designation, which expedites the development and review of drugs that are intended to treat a serious condition when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapies. It also received Orphan Drug designation.
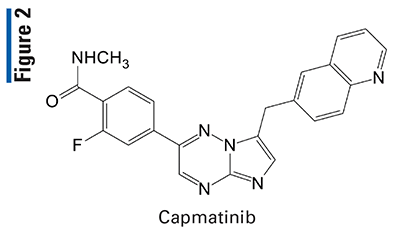
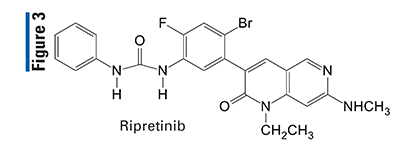
Pharmacology and Pharmacokinetics5,6: Ripretinib is a dihydronaphthyridine-3-phenylurea (FIGURE 3) designed to inhibit KIT proto-oncogene receptor tyrosine kinase and platelet-derived growth factor receptor A (PDGFRA) kinase, including wild-type, primary, and secondary mutations. Ripretinib also inhibits other kinases in vitro, such as PDGFRB, TIE2, VEGFR2, and BRAF. Inhibition of these kinases has been shown to block the proliferation of a number of cancers in which the heterogeneity of drug-resistant KIT mutations is a major therapeutic challenge, including GIST.
Ripretinib is well absorbed upon oral administration, giving peak plasma levels in 4 hours. It is primarily metabolized by CYP3A4 and to a lesser extent by CYP2C8 and CYP2D6, giving rise to an active metabolite designated as DP-5439. Both ripretinib and its active metabolite have large volumes of distribution (>300 L) and are highly bound by plasma proteins (>99%). The mean half-lives of ripretinib and DP-5439 are approximately 15 hours and 18 hours, respectively. Approximately 34% of the oral dose is excreted in feces as ripretinib and 6% as DP-5439, with only a small fraction of the parent drug and metabolite excreted in the urine. Based on this clearance profile, no dose adjustment is recommended in patients with mild hepatic impairment (total bilirubin £ULN and AST >ULN or total bilirubin 1 to 1.5 × ULN and any AST). A recommended dosage of ripretinib has not been established for patients with moderate or severe hepatic impairment.
Adverse Reactions and Drug Interactions5,6: The most common side effects (>20%) reported with ripretinib include alopecia, fatigue, nausea, abdominal pain, constipation, myalgia, diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome, and vomiting. Laboratory abnormalities (³4%), including increased lipase and decreased phosphate, were also observed. Ripretinib also may cause serious side effects, including skin cancer, hypertension, and cardiac dysfunction (decreased ejection fraction). Healthcare providers should routinely check for symptoms and signs of these and other risks. Also, since impaired wound healing may occur with this drug, ripretinib should be withheld for at least 1 week prior to surgery and at least 2 weeks post-surgery. Ripretinib may cause harm to a developing fetus or a newborn baby. Pregnant women, as well as females and males of reproductive potential, should be advised of these risks and to use effective contraception during treatment and for 1 week after the last dose. Patients should be advised not to breastfeed while taking ripretinib.
The use of strong CYP3A inducers should be avoided in patients on ripretinib therapy since they may compromise efficacy. Also, patients taking strong CYP3A4 inhibitors may experience increased exposure of ripretinib and its active metabolite (DP-5439), which may increase the risk of adverse reactions, requiring more frequent monitoring.
Dosage and Administration5,6: Ripretinib is supplied as 50-mg tablets for oral administration. The recommended dosage is 150 mg orally once daily, with or without food, until disease progression or unacceptable toxicity occurs. If adverse reactions occur, doses should be reduced to 100 mg orally once daily; dosing should be permanently discontinued in patients who are unable to tolerate 100 mg orally once daily. Ripretinib should be taken at the same time each day, and tablets should be swallowed whole. Patients should be advised to take a missed dose if less than 8 hours have passed since the missed scheduled dose. An additional dose should not be taken if vomiting occurs after taking the drug, but therapy continued with the next scheduled dose.
Avapritinib (Ayvakit, Blueprint Medicines)
Indication and Clinical Profile7,8: Avapritinib is specifically indicated for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GISTs) harboring a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation, including PDGFRA D842V mutations. GISTs arise in the smooth muscle pacemaker interstitial cell of Cajal, and their behavior is driven by mutations in the KIT gene (85%), PDGFRA gene (10%), or BRAF kinase (rare). Avapritinib is the first therapy approved for patients with GIST harboring a PDGFRA exon 18 mutation. The FDA granted this application priority review and Breakthrough Therapy designation, as well as Orphan Drug designations.
Avapritinib approval was based on results from the phase I NAVIGATOR clinical trial, as well as combined safety results from multiple clinical trials. The NAVIGATOR trial was a multicenter, single-arm, open-label trial enrolling 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 patients with PDGFRA D842V mutations. The trial initially enrolled patients at a starting dose of 400 mg orally once daily, but this was later reduced to 300 mg orally once daily due to toxicity. Patients received avapritinib until disease progression or unacceptable toxicity occurred. The major efficacy outcome measure was overall response rate (ORR) based on disease assessment by independent radiologic review using modified Response Evaluation Criteria in Solid Tumor 1.1 criteria. An additional efficacy outcome measure was response duration. For patients harboring a PDGFRA exon 18 mutation, the ORR was 84%, with 7% complete responses and 77% partial responses. For the subgroup of patients with PDGFRA D842V mutations, the ORR was 89%, with 8% complete responses and 82% partial responses. The median response duration was not reached with a median duration of follow-up for all patients of 10.6 months (range, 0.3–24.9 months); 61% of the responding patients with exon 18 mutations had a response lasting 6 months or longer (31% of patients with an ongoing response were followed for less than 6 months).
Pharmacology and Pharmacokinetics7,8: Avapritinib is a potent pyrazolopyrrole tyrosine kinase inhibitor (FIGURE 4 ) that targets PDGFRA and PDGFRA D842 mutants as well as multiple KIT exon 11, 11/17, and 17 mutants. Certain mutations in PDGFRA and KIT can result in the autophosphorylation and constitutive activation of these receptors, contributing to tumor cell proliferation. Other potential targets for avapritinib include wild-type KIT, PDGFRB, and CSFR1. In in-vitro cellular assays, avapritinib inhibited the autophosphorylation of KIT D816V and PDGFRA D842V, mutants associated with resistance to approved kinase inhibitors, with IC50 of 4 nM and 30 nM, respectively. Avapritinib also had antitumor activity in mice implanted with an imatinib-resistant patient-derived xenograft model of human GIST with activating KIT exon 11/17 mutations.
Avapritinib is well absorbed, producing peak plasma concentrations in 2 to 4 hours (Tmax). The maximum plasma concentration (Cmax) is increased by 59% and the AUC0-INF increased by 29% when avapritinib is taken with a high-calorie, high-fat meal. Steady state concentrations of the drug are reached 15 days following daily dosing. Avapritinib is highly bound by plasma proteins (98.8%) and has a mean apparent volume of distribution of 1200 L. It is primarily metabolized by oxidation by CYP3A4 and to a lesser extent by CYP2C9, and by glucuronidation to yield two primary metabolites, M690 (hydroxyl-glucuronide; 35%) and M499 (oxidative deamination; 14%). The mean plasma elimination half-life of avapritinib ranges from 32 to 57 hours. The drug and its metabolites are eliminated primarily in feces (70%).
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions (incidence ³20%) in patients who received avapritinib were edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. Depending on the severity of these reactions, avapritinib may be continued at the same dose, withheld, and then resumed at the same or reduced dose, or permanently discontinued.
Intracranial hemorrhage (e.g., subdural hematoma, intracranial hemorrhage, and cerebral hemorrhage) occurred in 1% of trial patients with GIST and overall in 3% of the 335 patients who received avapritinib. Avapritinib should be withheld for grade 1 or 2 reactions of this type until resolution and then resumed at a reduced dose. The drug should be permanently discontinued for recurrent grade 1 or 2 reactions or first occurrence of grade 3 or 4 reactions. Also, avapritinib may cause fetal harm, so females of reproductive potential and males with female partners of reproductive potential should be advised of the potential risk to a fetus and to use effective contraception. Patients using avapritinib should be advised not to breastfeed.
Based on its metabolic profile, coadministration of avapritinib should be avoided with strong and moderate CYP3A inhibitors; if coadministration cannot be avoided, the dose of avapritinib should be reduced. Coadministration of avapritinib should be avoided with strong and moderate CYP3A inducers to prevent loss of efficacy.
Dosage and Administration7,8: Avapritinib is supplied as 100-mg, 200-mg, and 300-mg tablets for oral administration. The recommended dosage is 300 mg orally once daily on an empty stomach, at least 1 hour before or 2 hours after a meal. Missed dose should not be made up within 8 hours of the next scheduled dose. Also, if vomiting occurs after administration, an additional dose should not be taken, but the patient should continue with the next scheduled dose. Treatment should be continued until disease progression or unacceptable toxicity occurs.
No dose adjustment is recommended for patients with mild or moderate renal impairment (creatinine clearance [CrCl] 30 to 89 mL/min). No dose recommendation has been established for patients with severe renal impairment (CrCl 15 to 29 mL/min) or end-stage renal disease (CrClr <15 mL/min). No dose adjustment is recommended for patients with mild or moderate hepatic impairment. Dose recommendations for avapritinib in severe hepatic impairment have not been established.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
REFERENCES
1. Zeposia (ozanimod) package insert. Summit, NJ: Celgene Corporation; March 2020.
2. Cohen JA, Comi G, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021-1033.
3. Tabrecta (capmatinib) package insert. East Hanover, N.J: Novartis Pharmaceuticals Corporation; May 2020.
4. Wolf J, Seto T, Han J, et al. Capmatinib in METex14-mutated advanced non-small cell lung cancer (NSCLC): efficacy data from the phase II GEOMETRY mono-1 study. Abstract 9004. Presented at: 2019 ASCO Annual Meeting; May 31-June 4, 2019; Chicago, IL.
5. Qinlock (ripretinib) package insert. Waltham, MA: Deciphera Pharmaceuticals, LLC; May 2020.
6. Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;21(7):923-934.
7. Ayvakit (avapritinib) package insert. Cambridge, MA: Blueprint Medicines Corporation; 2020.
8. Heinrich MC, Jones RL, von Mehren M, et al, Clinical activity of avapritinib in >fourth-line (4L+) and PDGFRA Exon 18 gastrointestinal stromal tumors (GIST) [abstract]. J Clin Oncol. 2019;37(15) suppl.
To comment on this article, contact rdavidson@uspharmacist.com.



