US Pharm. 2022;47(10):HS2-HS9.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2021–2022 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of an NME’s therapeutic profile are not detected in premarketing clinical studies and emerge after the drug is used in the broader population. For example, previously unreported adverse reactions become apparent for some NMEs within several years of their marketing. Some NMEs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers an introduction to some new drugs, it is essential that practitioners be aware of changes in their therapeutic profiles as reported in the pharmaceutical literature and by patients.
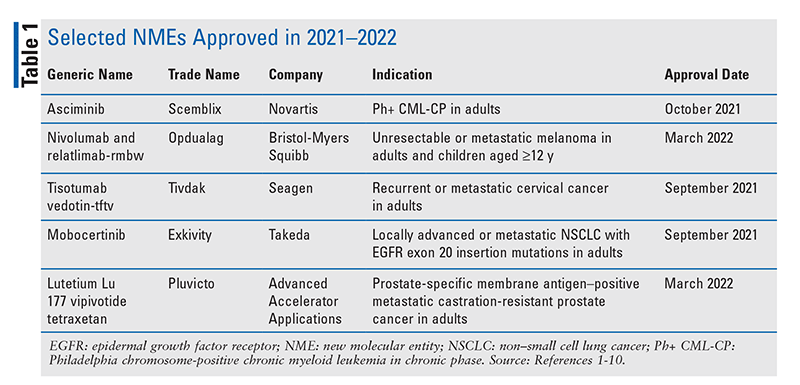
Asciminib (Scemblix, Novartis)
Indication and Clinical Profile1: Asciminib, a kinase inhibitor, received accelerated approval from the FDA for treatment of adult patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase (Ph+ CML-CP) previously treated with two or more tyrosine kinase inhibitors (TKIs). It also received standard approval for treatment of adult patients with Ph+ CML-CP with the T315I mutation.
Approval was based on results from the phase III ASCEMBL trial as well as a phase I trial in patients with Ph+ CML-CP with the T315I mutation (Ph+ CML-CP/T315I). In the phase I trial, 45 patients with Ph+ CML-CP/T315I received asciminib 200 mg twice daily and continued treatment until unacceptable toxicity or treatment failure. Major molecular response (MMR) was achieved by 24 weeks in 42% of patients, and MMR was achieved by 96 weeks in 49%. The median treatment duration was 108 weeks.
ASCEMBL included patients with Ph+ CML-CP who had experienced resistance or intolerance to at least two TKIs. Patients were randomized in a 2:1 ratio and stratified according to major cytogenetic response status to the experimental arm (asciminib 40 mg orally twice daily) or the active comparator arm (bosutinib 500 mg orally once daily). At 24 weeks, asciminib nearly doubled the MMR rate versus bosutinib (25% vs. 13%). The proportion of patients who discontinued treatment because of adverse reactions was more than three times lower in the asciminib arm versus the bosutinib arm (7% vs. 25%).
Pharmacology and Pharmacokinetics1,2: Asciminib (FIGURE 1) is an ABL/BCR-ABL1 TKI. Unlike other BCR-ABL inhibitors, such as imatinib, asciminib does not bind to the adenosine triphosphate–binding site on the active site of the enzyme; instead, it binds allosterically to the ABL myristoyl pocket. As a result, asciminib is active against wild-type BCR-ABL1 as well as multiple mutant drug-resistant forms of the kinase, including the T315I mutation.
Asciminib demonstrates a slight nonlinear exposure (AUC and maximum concentration of drug [Cmax]) at steady state within the dosing range of 10 mg to 200 mg. High-fat meals reduced the AUC and Cmax, and hepatic and renal impairment increased AUC extrapolated to infinity and Cmax. Steady-state volume of distribution is 151 L, and total apparent clearance is estimated at 6.7 L/h for 40 mg twice daily and 80 mg once daily and 4.1 L/h for 200 mg twice daily. The terminal elimination half-life ranges from 5.5 hours at 40 mg twice daily to 9 hours at 200 mg twice daily. Asciminib is metabolized by CYP3A4-mediated oxidation and UGT2B7- and UGT2B17-mediated glucuronidation.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions (³20%) reported with asciminib were upper respiratory tract infections, musculoskeletal pain, fatigue, nausea, rash, and diarrhea. The most common laboratory abnormalities (³20%) were decreased platelet, hemoglobin, and neutrophil counts and increased triglyceride, creatine kinase, alanine aminotransferase, lipase, and amylase levels.
No contraindications have been reported. Warnings and precautions include myelosuppression, pancreatic toxicity, hypertension, hypersensitivity, cardiovascular toxicity, and embryo-fetal toxicity. As a CYP3A4 substrate and inhibitor, asciminib should be used with caution with strong CYP3A4 inhibitors. Asciminib also inhibits CYP2C9 and P-glycoprotein. Asciminib should not be used concomitantly with an itraconazole oral solution containing hydroxypropyl-beta-cyclodextrin, which decreases asciminib’s efficacy.
Dosage and Administration1: Asciminib is available in tablets of 20 mg and 40 mg. The recommended dosage in patients with Ph+ CML-CP previously treated with two or more TKIs is 80 mg orally once daily or 40 mg orally twice daily. Recommended dosing for patients with Ph+ CML-CP with the T315I mutation is 200 mg orally twice daily. Asciminib should be taken without food, and food consumption should be avoided for at least 2 hours before and 1 hour after administration.
Nivolumab and Relatlimab-rmbw (Opdualag, Bristol-Myers Squibb)
Indication and Clinical Profile3,4: The combination drug containing nivolumab and relatlimab-rmbw was approved for treatment of unresectable or metastatic melanoma in adult and pediatric patients aged 12 years or older. Efficacy was established in Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma (RELATIVITY-047), a randomized, double-blind, phase II/III trial. RELATIVITY-047 compared the combination of nivolumab plus relatlimab with nivolumab monotherapy with regard to the primary outcome of progression-free survival (PFS). PFS was defined as the time between the date of randomization and the date of first documented tumor progression or death due to any cause, whichever occurred first. Secondary outcomes included overall survival and overall response rate. Median PFS was 10.1 months in the nivolumab-and-relatlimab arm versus 4.6 months for the monotherapy arm. The hazard ratio for progression or death was 0.75. The investigators concluded that inhibition of lymphocyte activation gene-3 (LAG-3) and programmed death receptor-1 (PD-1) provided a greater benefit with regard to PFS than inhibition of PD-1 alone in patients with previously untreated metastatic or unresectable melanoma.
Pharmacology and Pharmacokinetics3,4: This agent is a combination of nivolumab (a PD-1–blocking human immunoglobulin [Ig] G4 antibody) and relatlimab (a LAG-3–blocking human immunoglobulin IgG4 antibody). Nivolumab blocks the interaction of ligands PD-L1 and PD-L2 with their PD-1 receptor, reducing PD-1 pathway–mediated inhibition of the immune response, including the antitumor immune response. Relatlimab binds to the LAG-3 receptor, blocking interaction with its ligand, including major histocompatibility complex II; this action reduces LAG-3 pathway–mediated inhibition of the immune response. The combined actions of nivolumab and relatlimab increase T-cell activation to a greater extent than either antibody alone.
Administration of the recommended dosage of relatlimab resulted in a mean maximum concentration of 62.2 mcg/mL and an average concentration of 28.8 mcg/mL, whereas nivolumab resulted in a mean maximum concentration of 187 mcg/mL and an average concentration of 94.4 mcg/mL. The mean volume of distribution at steady state was 6.6 L for relatlimab and 6.6 L for nivolumab. Relatlimab’s clearance and half-life are 5.5 mL/h and 26.2 days, respectively, and correlate well with those for nivolumab (7.6 mL/h and 26.5 days, respectively). Variations in age, sex, race, mild or moderate renal impairment, and mild or moderate hepatic impairment had no clinically important effect on the clearance of nivolumab and relatlimab. The effects of severe renal or hepatic impairment on the pharmacokinetics of nivolumab and relatlimab are unknown.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (³20% of patients) reported with nivolumab and relatlimab-rmbw were musculoskeletal pain, fatigue, rash, pruritus, and diarrhea. Other adverse effects (>10%) included vitiligo, hypothyroidism, decreased appetite, nausea, cough, and headache. The most commonly reported laboratory abnormalities (³20%) were decreased hemoglobin, lymphocytes, and sodium and increased aspartate transaminase and alanine transaminase.
There are no contraindications to nivolumab and relatlimab-rmbw; however, warnings and precautions include severe or fatal immune-mediated adverse reactions, infusion-related reactions, complications of allogenic hematopoietic stem-cell transplantation, and embryo-fetal toxicity. This agent should not be used in pregnant women, as it has been shown to cause fetal harm. There are no available data on nivolumab and relatlimab-rmbw in pregnant women to evaluate a drug-associated risk.
Dosage and Administration3: This agent is supplied as a sterile, preservative-free solution of 240 mg nivolumab and 80 mg relatlimab per 20 mL (12 mg and 4 mg/mL) in single-dose vials. The recommended dosage for adults and pediatric patients aged 12 years or older weighing at least 40 kg is 480 mg nivolumab and 160 mg relatlimab administered IV every 4 weeks until disease progression or unacceptable toxicity. Nivolumab and relatlimab-rmbw may be administered diluted or undiluted. When diluted, this agent may be mixed with 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP, with a concentration range of nivolumab 3 mg/mL to 12 mg/mL and relatlimab 1 mg/mL to 4 mg/mL in adults and pediatric patients aged 12 years or older weighing at least 40 kg. The maximum infusion volume is 160 mL. Nivolumab and relatlimab-rmbw should be administered over 30 minutes through an IV line containing a sterile, nonpyrogenic, low-protein-binding, in-line polyethersulfone, nylon, or polyvinylidene filter (pore size: 0.2-1.2 µm). No dosage modifications are recommended.
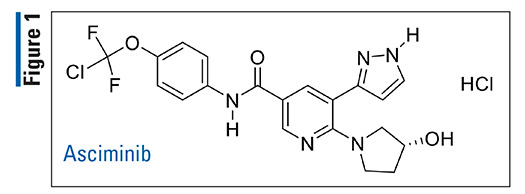
Tisotumab vedotin-tftv (Tivdak, Seagen)
Indication and Clinical Profile5,6: Tisotumab vedotin-tftv received approval for treatment of recurrent or metastatic cervical cancer in adults with disease progression on or after chemotherapy. Efficacy was established via innovaTV 204, a multicenter, open-label, single-arm, phase II trial conducted in Europe and the United States. The trial included patients aged 18 years or older who had recurrent or metastatic cervical cancer with squamous-cell, adenocarcinoma, or adenosquamous histology; had progressive disease during or after doublet chemotherapy plus bevacizumab, if eligible; had received no more than two previous systemic regimens for recurrent or metastatic cervical cancer; had measurable disease based on Response Evaluation Criteria in Solid Tumors (RECIST); and had an Eastern Cooperative Oncology Group performance status of 0 or 1.
The primary endpoint of the trial was confirmed objective response rate (ORR); secondary endpoints were duration of response, time to response, and progression-free survival. Patients received tisotumab vedotin-tftv 2 mg/kg (up to a maximum of 200 mg) IV every 3 weeks until independent review committee (IRC)–verified progressive disease as per RECIST (v1.1) criteria or unacceptable toxicity. Dose modifications were allowed for management of adverse events. An estimated confirmed ORR of 21% to 25% was used to determine a sample size of 100 patients, and patients were included for analysis if they received at least one dose of drug. All but one of 102 patients received at least one dose. The IRC-assessed confirmed ORR was 24%. Seven patients had a complete response and 17 patients had a partial response.
Pharmacology and Pharmacokinetics5,6: Tisotumab vedotin-tftv (FIGURE 2) is a tissue factor (TF)–directed antibody drug conjugate (ADC) consisting of an anti-TF IgG1-kappa antibody conjugated to monomethyl auristatin E (MMAE), a microtubule-disrupting agent. The anticancer activity of tisotumab vedotin-tftv is believed to involve the binding of ADC to TF-expressing cancer cells. This binding activity results in the release of MMAE, which inhibits actively dividing cells. In vitro, tisotumab vedotin-tftv also mediates antibody-dependent cellular phagocytosis as well as antibody-dependent cellular cytotoxicity.
Administration of one 3-week cycle of tisotumab vedotin-tftv 2 mg/kg resulted in peak mean concentrations of 40.8 mcg/mL near the end of the infusion. Unconjugated MMAE resulted in peak mean concentrations of 5.91 ng/mL 2 to 3 days after tisotumab vedotin-tftv dosing. Metabolism of the drug conjugate results in small peptides, amino acids, unconjugated MMAE, and unconjugated MMAE-related catabolites. Unconjugated MMAE is metabolized primarily by CYP3A4. The terminal half-life of tisotumab vedotin-tftv is 4.04 (range: 2.26-7.25) days.
Adverse Reactions and Drug Interactions5,6: The most common (³25%) adverse reactions were fatigue, nausea, diarrhea, peripheral neuropathy, alopecia, rash, epistaxis, vascular hemorrhage, conjunctival adverse reactions (conjunctivitis, conjunctival abrasion, conjunctival erosion, conjunctival hyperemia, conjunctival scare, noninfective conjunctivitis, ocular hyperemia, and conjunctival hemorrhage), and dry eye. Tisotumab vedotin-tftv has no contraindications; however, warnings include ocular adverse reactions, peripheral neuropathy, hemorrhage, pneumonitis, and embryo-fetal toxicity. Premedication and required eye care should be adhered to in order to reduce the risk of ocular adverse reactions. Recommendations for eye care include baseline and periodic ophthalmic examination and the use of topical corticosteroid eye drops, topical ocular vasoconstrictor drops, cold packs, topical lubricating eye drops, and contact lenses.
Drug interactions with tisotumab vedotin-tftv involve the metabolism of unconjugated MMAE via CYP3A4. The concomitant use of tisotumab vedotin-tftv with strong CYP3A4 inhibitors may increase unconjugated MMAE exposure, which may heighten the risk of adverse reactions.
Dosage and Administration5: Tisotumab vedotin-tftv is supplied as a single-dose 40-mg vial for reconstitution. The recommended dosage for patients weighing 100 kg or more is 2 mg/kg, up to a maximum of 200 mg, every 3 weeks until disease progression or unacceptable toxicity. The drug should be administered as an infusion over 30 minutes through an IV line containing a 0.2-µm in-line filter. The dosage may be reduced to 1.3 mg/kg for the first reduction and 0.9 mg/kg for the second reduction, and therapy should be discontinued permanently in patients unable to tolerate 0.9 mg/kg. Once the single-dose vial is appropriately reconstituted, the calculated dose may be diluted with 5% Dextrose Injection, USP, 0.9% Sodium Chloride Injection, USP, or Lactated Ringer’s Injection, USP. The infusion bag should be of appropriate size to allow enough diluent to achieve a final tisotumab vedotin-tftv concentration of 0.7 mg/mL to 2.4 mg/mL. The patient should be pretreated with steroid and vasoconstrictor eye drops prior to administration. Cold packs should be applied (with the eyes fully covered) following the administration of eye drops, and the cold packs should remain in place throughout the infusion, changing as needed to ensure that the eye area remains cold.
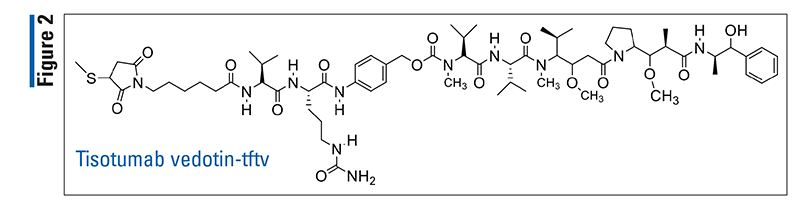
Mobocertinib (Exkivity, Takeda)
Indication and Clinical Profile7,8: Mobocertinib, a kinase inhibitor, is approved for treatment of locally advanced or metastatic non–small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations (as confirmed by an FDA-approved test) in adults whose disease has progressed on or after platinum-based chemotherapy. This indication was approved under accelerated approval based on overall response rate (ORR) and duration of response (DoR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Approval is based on results of a phase I/II trial of mobocertinib involving 114 patients with EGFR exon 20 insertion–positive NSCLC who had received prior platinum-based chemotherapy. Patients were treated with 160 mg once daily until disease progression or intolerable toxicity. The ORR was 28% per blinded independent central review; investigator-assessed ORR was 35%, with a median DoR of 17.5 months. Median overall survival was 24 months, and median progression-free survival was 7.3 months.
Pharmacology and Pharmacokinetics7,8: Mobocertinib (FIGURE 3) is a kinase inhibitor designed to selectively target EGFR exon 20 insertion mutations at lower concentrations than wild-type EGFR. In vitro, mobocertinib also inhibited the activity of other EGFR family members (HER2 and HER4) and one additional kinase (B lymphocyte kinase) at clinically relevant concentrations (half maximal inhibitory concentration values <2 nM).
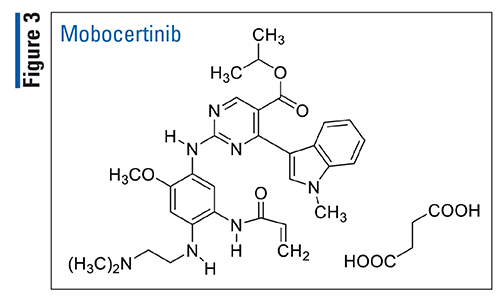
The mean bioavailability of mobocertinib is 37%, and the time to peak concentration (Tmax) is 4 hours. The drug has an apparent volume of distribution of about 3,500 L at steady state, and it is 99% bound by plasma proteins. Mobocertinib is metabolized primarily by CYP3A, giving rise to two active metabolites—AP32960 and AP32914—that are equipotent to the parent drug. Mobocertinib is excreted primarily in the feces (76%) as metabolites (with 6% as unchanged mobocertinib). Mean plasma elimination half-life is 18 hours at steady state. No clinically significant differences in pharmacokinetics were observed based on age, race, sex, body weight, mild or moderate renal impairment, or mild hepatic impairment. The effect of severe renal or hepatic impairment on kinetics was not determined.
Adverse Reactions and Drug Interactions7,8: Adverse reactions with mobocertinib include diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, and dry skin. Grade 3/4 laboratory abnormalities, including decreased lymphocytes, increased amylase, increased lipase, decreased potassium, decreased hemoglobin, increased creatinine, and decreased magnesium, have also been reported. The label includes a black box warning that mobocertinib can cause life-threatening heart rate–corrected QT (QTc) prolongation, including torsades de pointes, which can be fatal. Therefore, it is necessary to monitor QTc and electrolytes at baseline and periodically during treatment; monitoring should be conducted more frequently in patients with risk factors for QTc prolongation. Concomitant use of other drugs known to prolong the QTc interval or moderate to strong CYP3A inhibitors should be avoided with mobocertinib, as they may further prolong the QTc. Based on the severity of QTc prolongation, when it occurs, mobocertinib therapy should be withheld, reduced, or permanently discontinued.
Dosage and Administration7,8: Mobocertinib is supplied as capsules of 40 mg. The recommended dosage is 160 mg orally once daily until disease progression or unacceptable toxicity. The drug should be taken at the same time each day, with or without food. The capsules should be swallowed whole, not chewed, opened, or dissolved in liquids. If a dose is missed by more than 6 hours, that dose should be skipped and the next dose taken the following day at its regularly scheduled time. If a dose is vomited, an additional dose should not be taken; instead, the next dose should be taken as prescribed the following day.
Lutetium Lu 177 vipivotide tetraxetan (Pluvicto, Advanced Accelerator Applications)
Indication and Clinical Profile9,10: Lutetium Lu 177 vipivotide tetraxetan is indicated for treatment of prostate-specific membrane antigen (PSMA)–positive metastatic castration-resistant prostate cancer (mCRPC) in adults who have received androgen-receptor pathway inhibition and taxane-based chemotherapy. Because PSMA is highly expressed in >80% of PC patients, it is an important phenotypic biomarker for assessing the progression of metastatic PC. PC is the leading cause of cancer-related death in the United States; metastatic PC has a 5-year survival rate of <30%, and mCRPC patients who progress on multiple lines of therapy have limited treatment options.
Lutetium Lu 177 vipivotide tetraxetan is the first FDA-approved targeted radioligand therapy (RLT) for eligible mCRPC patients that combines a prostate-targeting compound (ligand) with a therapeutic radioisotope (radioactive particle). The FDA also approved Locametz, a gallium Ga 68 gozetotide radiolabeling imaging agent, for positron emission tomography of PSMA-positive lesions in adult patients with mCRPC. This agent can identify patients eligible for targeted treatment with lutetium Lu 177 vipivotide tetraxetan and locate areas in the body where PSMA tumors may have spread (e.g., soft tissue, lymph nodes, or bone).
Approval of lutetium Lu 177 vipivotide tetraxetan is based on results of the prospective, randomized, open-label, international, multicenter, phase III VISION trial, which included 831 patients. This study assessed the efficacy and safety of lutetium Lu 177 vipivotide tetraxetan (7.4 GBq administered by IV infusion every 6 weeks for a maximum of six cycles) plus investigator-chosen standard of care (SOC) in the investigational arm versus SOC in the control arm. Participants treated with lutetium Lu 177 vipivotide tetraxetan plus SOC had a 38% reduction in risk of death and a statistically significant reduction in risk of radiographic disease progression or death compared to SOC alone. In addition, 30% of patients with evaluable disease at baseline demonstrated an overall response (per RECIST 1.1) with lutetium Lu 177 vipivotide tetraxetan plus SOC, compared with 2% in the SOC-alone arm.
Pharmacology and Pharmacokinetics9,10: Lutetium Lu 177 vipivotide tetraxetan (FIGURE 4), as discussed previously, is a radioligand therapeutic agent. The active anticancer moiety is the radionuclide lutetium-177, which is linked to a targeting moiety that binds to PSMA, a transmembrane protein that is expressed in PC (including mCRPC). Upon binding to PSMA-expressing cells, the beta-minus emission from lutetium-177 delivers radiation to PSMA-expressing cells as well as to the surrounding cells; it also induces DNA damage, thereby triggering cell death.
Upon IV administration of lutetium Lu 177 vipivotide tetraxetan, the AUC is 52.3 ng.h/mL (31.4%) and the maximum blood concentration is 6.58 ng/mL (43.5%) at the recommended dosage. The drug’s volume of distribution is 123 L, and it is 60% to 70% bound by plasma proteins. Lutetium Lu 177 vipivotide tetraxetan is primarily eliminated renally, and the terminal elimination half-life of this agent is 41.6 hours.
Adverse Reactions and Drug Interactions9,10: Adverse reactions that are associated with the use of lutetium Lu 177 vipivotide tetraxetan include fatigue, dry mouth, nausea, anemia, decreased appetite, and constipation; laboratory abnormalities include decreased lymphocytes, hemoglobin, leukocytes, platelets, calcium, and sodium. The safety and efficacy of this agent have not been established in females, but based on its mechanism of action, it is likely to cause harm to an exposed fetus. Patients should be advised to refrain from sexual activity for 7 days following treatment; they should sleep in a bedroom separate from household contacts (3 days), children (7 days), or pregnant women (15 days). Male patients with female partners of reproductive potential should be advised to use effective contraception during treatment and for 14 weeks after the last dose.
Lutetium Lu 177 vipivotide tetraxetan is not a substrate, inhibitor, or inducer of any major cytochrome isozymes or transporters; therefore, the occurrence of metabolic and excretory drug interactions is not anticipated.
Dosage and Administration9,10: Lutetium Lu 177 vipivotide tetraxetan is a radiopharmaceutical, and it should be handled with appropriate safety measures in order to minimize radiation exposure. It is supplied as an injection for IV administration. The recommended dosage is 7.4 GBq (200 mCi) IV every 6 weeks for up to six doses, or until disease progression or unacceptable toxicity. It may be necessary to temporarily interrupt dosing (extending the dosing interval from every 6 weeks up to every 10 weeks), reduce the dose, or permanently discontinue treatment to manage adverse effects. If a treatment delay due to an adverse reaction persists for >4 weeks, lutetium Lu 177 vipivotide tetraxetan treatment should be discontinued. The dose may be reduced by 20% to 5.9 GBq (160 mCi) once; do not reescalate. If a patient has further adverse reactions that would require an additional dosage reduction, treatment must be discontinued.
REFERENCES
1. Scemblix (asciminib) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; October 2021.
2. Lexicomp Online. Asciminib: drug information. Waltham, MA: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
3. Opdualag (nivolumab and relatlimab-rmbw) package insert. Princeton, NJ: Bristol-Myers Squibb Co; March 2022.
4. Tawbi HA, Schadendorf D, Lipson EJ, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24-34.
5. Tivdak (tisotumab vedotin-tftv) package insert. Bothell, WA: Seagen Inc; January 2022.
6. Lexicomp Online. Tisotumab: drug information. Waltham, MA: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
7. Exkivity (mobocertinib) package insert. Lexington, MA: Takeda Pharmaceuticals America, Inc; September 2021.
8. Zhang SS, Zhu VW. Spotlight on mobocertinib (TAK-788) in NSCLC with EGFR exon 20 insertion mutations. Lung Cancer (Auckl). 2021;12:61-65.
9. Pluvicto (lutetium Lu 177 vipivotide tetraxetan) package insert. Millburn, NJ: Advanced Accelerator Applications USA, Inc; March 2022.
10. Neels OC, Kopka K, Liolios C, Afshar-Oromieh A. Radiolabeled PSMA inhibitors. Cancers (Basel). 2021;13(24):6255.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


